Abstract
Expansions of gene-specific DNA tandem repeats (TRs), first described in 1991 as a disease-causing mutation in humans, are now known to cause >60 phenotypes, not just disease, and not only in humans. TRs are a common form of genetic variation with biological consequences, observed, so far, in humans, dogs, plants, oysters, and yeast. Repeat diseases show atypical clinical features, genetic anticipation, and multiple and partially penetrant phenotypes among family members. Discovery of disease-causing repeat expansion loci accelerated through technological advances in DNA sequencing and computational analyses. Between 2019 and 2021, 17 new disease-causing TR expansions were reported, totaling 63 TR loci (>69 diseases), with a likelihood of more discoveries, and in more organisms. Recent and historical lessons reveal that properly assessed clinical presentations, coupled with genetic and biological awareness, can guide discovery of disease-causing unstable TRs. We highlight critical but underrecognized aspects of TR mutations. Repeat motifs may not be present in current reference genomes but will be in forthcoming gapless long-read references. Repeat motif size can be a single nucleotide to kilobases/unit. At a given locus, repeat motif sequence purity can vary with consequence. Pathogenic repeats can be “insertions” within nonpathogenic TRs. Expansions, contractions, and somatic length variations of TRs can have clinical/biological consequences. TR instabilities occur in humans and other organisms. TRs can be epigenetically modified and/or chromosomal fragile sites. We discuss the expanding field of disease-associated TR instabilities, highlighting prospects, clinical and genetic clues, tools, and challenges for further discoveries of disease-causing TR instabilities and understanding their biological and pathological impacts—a vista that is about to expand.
More than 30 years ago, in 1991, expansions of DNA tandem repeats (TRs) at particular loci were first shown to cause human diseases, termed repeat expansion diseases (Kremer et al. 1991; La Spada et al. 1991; Oberlé et al. 1991; Verkerk et al. 1991; Yu et al. 1991). After an initial period of successive identifications of similar trinucleotide repeat expansions (Pearson et al. 2005; López Castel et al. 2010), the rate of TR-associated disease discovery slowed as the limitations of technological methods reduced the ability to detect more complex pathogenic repeat expansions. However, recent technological advances in both DNA sequencing techniques and computational analysis have again increased speed of discovery, with 17 new disease-causing and risk-associated TR expansions being published between 2019 and 2021 (Fig. 1; Table 1; Corbett et al. 2019; Cortese et al. 2019; Demaerel et al. 2019; Florian et al. 2019; Ishiura et al. 2019; LaCroix et al. 2019; Sone et al. 2019; Tian et al. 2019; van Kuilenburg et al. 2019; Yeetong et al. 2019; Katsumata et al. 2020; Ruggieri et al. 2020; Pagnamenta et al. 2021; Yeetong et al. 2021). The most recently identified mutations were “difficult sequences” for conventional techniques, caused either by GC-rich repeat-motif sequences that are difficult to amplify by PCR (Ishiura et al. 2019; LaCroix et al. 2019; Sone et al. 2019; Tian et al. 2019; van Kuilenburg et al. 2019), or by repeat sequence motifs within TR stretches that are not found within the reference genome (Sato et al. 2009; Seixas et al. 2017; Ishiura et al. 2018; Corbett et al. 2019; Cortese et al. 2019; Demaerel et al. 2019; Florian et al. 2019; LaCroix et al. 2019; Yeetong et al. 2019; Katsumata et al. 2020; Ruggieri et al. 2020).
Overview of disease-associated repeat discovery by year, with colored inserts specifying the major technological breakthroughs that were used to make these discoveries. (AD) Alzheimer disease, (ALS/FTD) amyotrophic lateral sclerosis/frontotemporal dementia, (ASD) autism spectrum disorder, (BAFME) benign adult familial myoclonic epilepsy, (BD) bipolar disorder, (BPES) blepharophimosis, ptosis, and epicanthus inversus syndrome, (CANVAS) cerebellar ataxia, neuropathy, vestibular areflexia syndrome, (CCD) cleidocranial dysplasia, (CCHS) congenital central hypoventilation syndrome, (DBQD2) Desbuquois dysplasia 2, (DM) myotonic dystrophy, (DRPLA) dentatorubropallidoluysian atrophy, (EDM1) multiple epiphyseal dysplasia, (EIEE1) epileptic encephalopathy, early infantile, 1, (EPM1) epilepsy, progressive myoclonus-1, (FECD3) Fuchs endothelial corneal dystrophy-3, (FRDA) Friedreich's ataxia, (FSHD) facioscapulohumeral muscular dystrophy, (FXTAS) fragile X ataxia/tremor syndrome, (GD) glutaminase deficiency, (HDL2) Huntington disease-like 2, (HFG) hand-foot-genital syndrome, (HPE5) holoprosencephaly 5, (LOAD) late-onset Alzheimer disease, (MJD) Machado-Joseph disease, (NIID) neuronal intranuclear inclusion disease, (OPDM) oculopharyngodistal myopathy, (OPMD) oculopharyngeal muscular dystrophy, (OPML) oculopharyngeal myopathy with leukoencephalopathy, (PSACH) pseudoachondroplasia, (RCPS) Richieri-Costa-Pereira syndrome, (SBMA) spinal bulbar muscular atrophy, (SCA) spinocerebellar ataxia, (SMD) skeletal muscle disease, (SPD1) synpolydactyly-1, (SCZ) schizophrenia, (XPD) X-linked dystonia-parkinsonism, (22q11DS) 22q11 deletion syndrome. It has been concluded that FAME, BAFME, FEME, FCTE, and ADCME are the same clinical entity even if genetically heterogeneous—we use the acronym BAFME here as it is the most used acronym associated with the disease. The nonfolate-sensitive rare fragile sites FRA10B and FRA16B, caused by expanded AT-rich repeats, are not listed herein (see Table 1).
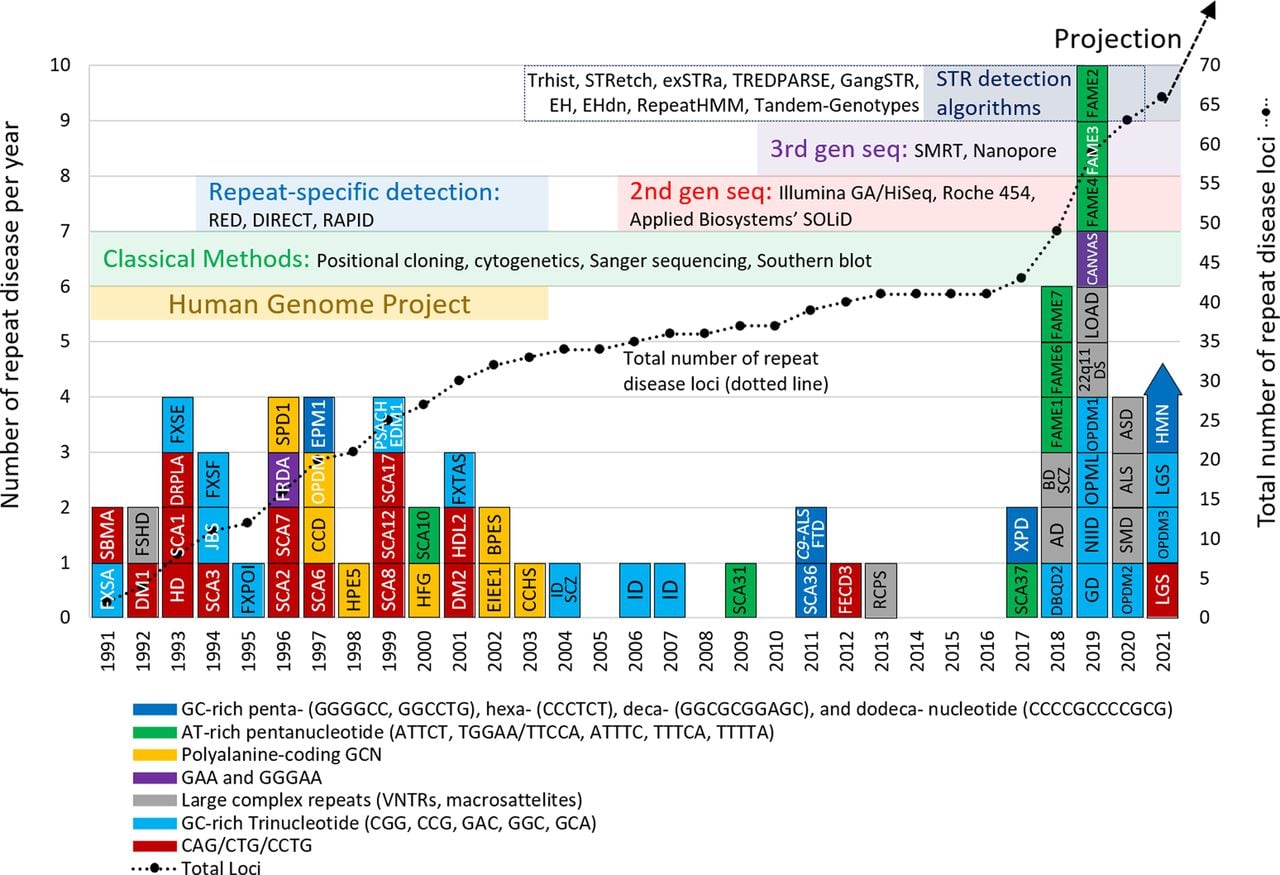
Recent repeat mutation identifications also highlight the importance of functional and clinical aspects of TR expansions in the discovery process. Expansions of TRs of the same repeat unit motifs can cause diseases with similar phenotypes independent of their genetic loci, supporting a gain-of-function pathogenesis hypothesis. For example, several SCAs caused by expansions of CAG repeat motifs present with similar motor phenotypes despite their expansions occurring within different genetic loci. More recently, careful analysis and categorization of clinical manifestations served as essential tools in recent discoveries where the same expansion mutations occurring at different genomic loci all resulted in benign adult familial myoclonic epilepsy (BAFME), also known as familial adult myoclonic epilepsy (FAME) (Ishiura et al. 2018, 2019; Corbett et al. 2019; Florian et al. 2019; Yeetong et al. 2019). Further, other new discoveries also reminded us of relatively underrecognized loss-of-function mechanisms which may precipitate pathogenesis. In the cases of Desbuquois dysplasia 2 (DBQD2; also known as Baratela-Scott syndrome) and glutaminase deficiency (GD), expansions of GC-rich TR sequences in the promoter regions cause pathogenic transcriptional suppression (LaCroix et al. 2019; van Kuilenburg et al. 2019). Further exploration of suspected loss-of-function mechanisms may therefore be beneficial in understanding how repeat mutations elicit pathogenesis.

While there have been a number of excellent reviews on diseases associated with TR expansions, they are mainly focused on disease mechanisms (Hannan 2018; Rodriguez and Todd 2019). As such, considering the success of recent studies in the identification of new repeat disease motifs, the focus of this review is to highlight how DNA sequencing technologies and analytic approaches, coupled with clinical and biological assessment, facilitate repeat disease mutation discovery and our understanding of pathogenic mechanisms. We begin with a brief overview of the history of repeat disease gene discovery, with an emphasis on how these discoveries facilitated further discovery. Next, we will explore how new technologies are making more difficult sequences in the genome accessible and discuss the need for further development of analytical tools. Lastly, we will highlight how some of the recent findings identified relatively underrecognized clinical and mechanistic features of TR-expansion-related disorders, which should not be overlooked as future research aims to improve our understanding of repeat diseases and their underlying mechanisms. By covering these topics, we attempt to provide guidance for future investigations into TRs and their roles in physiological and disease processes through the integration of technology and biological understanding.
Part 1: Technological advances and repeat disease mutation discovery
Historical overview of disease-associated repeat expansion discovery
The initial discoveries in the early 1990s were trinucleotide repeats, namely a CGG repeat in the 5′ UTR of FMR1 (Kremer et al. 1991; Oberlé et al. 1991; Pieretti et al. 1991; Verkerk et al. 1991; Yu et al. 1991), a polyglutamine-coding CAG repeat in the AR gene (La Spada et al. 1991), and a CTG repeat in the 3′ UTR of DMPK (Aslanidis et al. 1992; Brook et al. 1992; Buxton et al. 1992; Fu et al. 1992; Harley et al. 1992; Mahadevan et al. 1992). Expansions of these repeats caused fragile X syndrome (FXS), spinal and bulbar muscular atrophy (SBMA), and myotonic dystrophy type 1 (DM1), respectively. It was later found that tetra- (Liquori et al. 2001), penta- (Matsuura et al. 2000; Sato et al. 2009), hexa- (DeJesus-Hernandez et al. 2011; Kobayashi et al. 2011; Renton et al. 2011), and dodeca- (Lafrenière et al. 1997; Virtaneva et al. 1997) nucleotide repeat expansions in intronic or promoter regions can also result in other human diseases (Fig. 1; Table 1).
Some of these repeat disorders exhibited a peculiar set of phenomena from the viewpoint of conventional Mendelian inheritance: “anticipation” (where successive generations show earlier disease onset and more severe phenotypes), variable disease phenotypes among family members (Bell 1941; Martin and Bell 1943; Sherman et al. 1984 1985; Höweler et al. 1989; Sutherland et al. 1991; Harper et al. 1992; Mandel 1993; Pearson et al. 2005) and, for some diseases like SCA8, also presenting reduced penetrance (Koob et al. 1999). These clinical phenomena, luckily, did not hinder repeat mutation discovery, but were instead viewed as a central characteristic of TR expansions, and this clinical awareness led to more and more similar mutations being identified (Kawaguchi et al. 1994; Koide et al. 1994; Nagafuchi et al. 1994; Pearson et al. 2005).
Initially, expansion mutations were discovered through positional cloning (Fig. 1), and cytogenetic mapping—for example, the identification of the CGG expansion mutation responsible for the cytogenetic fragile site, FRAXA, which until then had been the main diagnostic marker of FXS (Lubs 1969). While detailed coverage is beyond the scope of this review, an appreciation of the approaches used is relevant. Among a variety of competing potential theories (not all covered here), one was the hypothesis that an unstable repeat sequence would be the cause of the fragile site FRAXA and disease FXS. Those initial suspicions, hypothesizing the involvement of an unstable amplified repeat tract, were based upon the biology of chromosomal fragile site induction and the puzzling genetics of the disease (Sutherland et al. 1985; Nussbaum et al. 1986; Hori et al. 1988). Among the first experimental evidence supporting the involvement of an unstable DNA sequence were cytogenetic observations of chromosomal instability at the fragile site in rodent-human somatic cell hybrids; a reagent subsequently cloned, sequenced, and localized cytogenetically (FISH) the causative CGG expansion mutation (Warren et al. 1987). Warren and colleagues, citing the repeat-hypothesis proposed in 1985 (Sutherland et al. 1985; Ledbetter et al. 1986; Nussbaum et al. 1986; Hori et al. 1988), concluded “… that the fragile X site is a reiterated DNA sequence of variable length, the longest length being found in fully penetrant males and the shortest in phenotypically normal individuals… Fragility in this region of the X has been shown to support this model in that normal, transmitting, and affected male X chromosomes (in somatic cell hybrids) show increasing frequencies of fragility… [T]he observation of reduced chromosome fragility at the translocation junctions lends support for the model of the fragile X site as a reiterated DNA sequence.” In 1991, the concept of genetic instability was further supported by variably slow-migrating DNAs on Southern blots—suspected as amplified repeats (Oberlé et al. 1991; Yu et al. 1991) and soon after revealed as a CGG expansion (Fu et al. 1991; Kremer et al. 1991; Pieretti et al. 1991; Verkerk et al. 1991). This was possible through using cytogenetics/FISH, coupled with somatic cell hybrids for FRAXA breakpoint mapping, Alu-PCR, and positional cloning, which together permitted identification of the CGG expansion in FMR1 (Warren et al. 1987; Bell et al. 1991; Heitz et al. 1991; Kremer et al. 1991; Vincent et al. 1991). Cytogenetics/FISH and molecular genetics are still required to validate the molecular mapping of fragile sites (Warren et al. 1987; Bell et al. 1991; Heitz et al. 1991; Kremer et al. 1991; Vincent et al. 1991). For specific details of FRAXA/CGG/FMR1 discoveries, we refer readers to a focused review, published during that early time (Oostra and Verkerk 1992). Indeed, it seems that the advances of the fragile X research, discovering a repeat expansion as the genetic cause for a disease with unusual inheritance patterns (Sherman paradox), incomplete penetrance, and strong parent-of-origin effects, paved the way for repeat-centered efforts for many of the other diseases. The localization of the mutant regions of other repeat diseases involved the use of many mapping techniques, including radiation-reduced hybrids, flow-sorted chromosome libraries, CpG island screens, exon trapping, exon amplification, and use of cosmid/yeast artificial chromosome libraries (La Spada et al. 1991; Aslanidis et al. 1992; Buxton et al. 1992; The Huntington Disease Collaborative Research Group 1993). The probing of positionally mapped disease regions for suspected repeat tract length variations led to the discovery of many of the other diseases that similarly showed unusual inheritance patterns and parent-of-origin effects. It was predicted that the mutation causing DM1, which showed strong genetic anticipation, similar to the Sherman paradox of FXS, could be caused by an unstable repeat (Höweler et al. 1989; Sutherland et al. 1991). Following the discovery of the DM1 mutation as an expanded CTG repeat based upon its tight association with genetic anticipation, it was predicted that HD and SCAs (known then as olivopontocerebellar ataxias) would be caused by gene-specific repeat expansions (Caskey et al. 1992; Harper et al. 1992). Following the explanation of HDs genetic anticipation by a CAG expansion (Snell et al. 1993; Trottier et al. 1994), the connection was solidified, and it was predicted that SCAs, bipolar disorder/schizophrenia, and non-FXS linked autism could also be caused by repeat expansions (Pulst et al. 1993; Ross et al. 1993). Each of these predictions, to some degree, turned out to be true for numerous SCAs, associatively for at least one form of bipolar disorder/schizophrenia (CACNA1C) (Song et al. 2018), and most recently, autism spectrum disorder (ASD) (Trost et al. 2020). It is notable that SBMA does not show obvious genetic anticipation nor high levels of repeat instability, and the discovery of the CAG expansion in the androgen receptor in affected families was a further extension of the already known polymorphism of the repeat in the unaffected population (Lubahn et al. 1988; Tilley et al. 1989; Edwards et al. 1991, 1992; La Spada et al. 1991).
While these initial discoveries used technologies that were mostly not repeat-specific, following the discoveries of several CAG/CTG expansions, a series of methodological protocols was developed to detect expansions of this trinucleotide repeat without knowledge of their genomic loci: Repeat Expansion Detection (RED) (Schalling et al. 1993), Direct Identification of Repeat Expansion and Cloning Technique (DIRECT) (Sanpei et al. 1996), and Repeat Analysis, Pooled Isolation, and Detection of expanded trinucleotide repeat clones (RAPID) (Koob et al. 1998). These protocols were based on completely novel ideas in the era of positional cloning and were used to identify several new disease loci caused by unstable repeat expansions: RED brought about the discoveries of spinocerebellar ataxia type 7 (SCA7) (Lindblad et al. 1996), SCA12 (Holmes et al. 1999), Huntington disease-like 2 (HDL2) (Holmes et al. 2001; Margolis et al. 2001) and CTG18.1 (Breschel et al. 1997); DIRECT led to the identification of the ATXN2 mutation (Sanpei et al. 1996) and RAPID to the SCA8 repeat expansion (Koob et al. 1998). It should also be noted that the discoveries of the CAG mutations causative for SCA2 and SCA7 were immensely facilitated by detection of polyQ aggregates with a monoclonal antibody, which predicted expansions in SCA2 through detection of expansions in extracts of SCA2 patient cells (Trottier et al. 1995).
The continuous discovery of new TR expansion mutations in the 1990s fully leveraged the power of the Human Genome Project, as the huge numbers of sequence-tagged site (STS) markers that became available enabled fine mapping of the disease loci. Today, locus mapping can be done with high-density SNP typing using microarrays (Gentalen and Chee 1999), which facilitates the completion of linkage analysis more rapidly than ever before. The reference sequence of the human genome and the variation database made “resequencing” approaches possible. Following this, the first decade of the 21st century witnessed rapid development of new DNA sequencing technologies, now called second-generation sequencing (or next-generation sequencing; NGS). Three of the first to be widely used were Illumina's GA/HiSeq Systems, 454 Life Sciences’ 454 System and Applied Biosystems’ Sequencing by Oligo Ligation Detection (SOLiD) (van Dijk et al. 2014). Together with the completion of draft human genome sequence, these high-throughput systems contributed to the high number of genomic variations discovered to result in various human phenotypes. The new sequencing technologies led to an increasing number of gene discoveries for Mendelian conditions from 2010 onward (Bamshad et al. 2019). However, due to limitations in analytical tools available to handle repeat sequences and the technical weaknesses associated with fidelity and processivity of DNA polymerases, it took another decade for new sequencing technologies to begin to enhance identifying disease-causing TR expansions (Fig. 1).
Bioinformatic algorithms are unleashing the potential of NGS for repeat disease discovery
Application of NGS approaches in this field was first published in 2011 in one of two papers that reported the discovery of the GGGGCC repeat expansion in the C9orf72 gene associated with familial amyotrophic lateral sclerosis/frontotemporal dementia (ALS/FTD) (Renton et al. 2011). Massive parallel paired-end sequencing by HiSeq 2000 permitted rapid data collection, but the expanded repeat was identified through manual inspection and realignment of the sequence data in the candidate region, which was only possible because the linkage block had been narrowed down to a region of 232 kb. Validation of the C9orf72 repeat expansion in 2011 required the use of Southern blotting (DeJesus-Hernandez et al. 2011), a method still required in 2020 for validation of repeat expansions (Trost et al. 2020). Even more recently, in a 2019 study, biallelic expansions of an (AAGGG)N repeat in the intron of RFC1 were identified as responsible for cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS) by a similar method—visual analysis of assembled short-read sequencing (SRS) data generated by HiSeq 4000 system within a 1.7-Mb candidate region (Cortese et al. 2019). Despite these success stories in the use of NGS, over this same time period, several repeat disease mutation discoveries still depended on conventional methods despite the technological advances. For example, the 2009 identification of the mutation that causes SCA31 was achieved solely by “traditional” methods: bacterial artificial chromosome (BAC)-based cloning, Sanger sequencing, and Southern blot, with targeted shotgun resequencing (Sato et al. 2009).
The techniques employed by the Human Genome Project notoriously struggled with TR stretches and, in fact, there are still regions with long TRs yet to be correctly assembled—for example, the classical satellite repeats I-IV (Miga 2015). Shorter simple tandem repeats (STR), with 1- to 6-bp motif units, were also underrecognized, with recent bioinformatic assessment showing that these STRs comprise 6.77% of the human genome—more than twice what was initially predicted (Shortt et al. 2020). To this day, repeats pose significant hurdles for even NGS, ranging from technical hurdles including difficulties of bacterial cloning, PCR amplification, and sequence read size limits, to computational hurdles including misalignment and omission of the repeat or flanks from the reference genome (LaCroix et al. 2019). The high GC-content of TRs, and of the deletion-prone regions in which they are often embedded, can account for hindering the identification of a disease-causing mutation, as highlighted by the recent discovery of a GC-rich 10-mer repeat with compound heterozygous deletions (Pagnamenta et al. 2021) and of biallelic deletions in the VWA1 gene (Deschauer et al. 2021). VWA1 had been identified as the disease-causing gene of neuromyopathy and loss-of-function supported by animal models, but the mutation remained veiled by technical hurdles. Similarly, the identification of repeat expansions in the CSTB gene of either a 15-mer, an 18-mer (Virtaneva et al. 1997), or a dodecamer (12-mer) minisatellite repeat expansion (Lafrenière et al. 1997; Lalioti et al. 1997). Further analysis confirmed a dodecamer composed exclusively of G and C residues (Lalioti et al. 1997). From this, it is clear that both first generation and NGS and analysis of NGS data struggle to detect TRs, with recent advances demonstrating that much more sophisticated bioinformatic tools are necessary for their detection (van der Sanden et al. 2021).
One major technical issue is that disease-associated expanded TR tracts (whose tracts are, for the most part, much shorter than those in satellite DNA) have been tough obstacles for Illumina's widely used HiSeq systems that depend on assembly of short-read sequences (typically paired 150-base reads). This is because reads filled entirely or partially with expanded simple repeat sequences cannot be assembled accurately and because sequences derived from TRs are associated with a higher sequencing error rate due to their low complexity and/or due to their high GC-content, thereby further promoting misassembly (Benjamini and Speed 2012).
One way to overcome this is to increase the sequencing read length so that flanking sequences are encompassed within the read to allow for simpler alignment (Ummat and Bashir 2014). This is especially needed after the 454 System—which had relatively long read lengths among second generation sequencers, reaching up to 1 kb—was discontinued. The recent discovery of CGG repeat expansions in neuronal intranuclear inclusion disease (NIID) was achieved with long-read whole-genome sequencing (WGS), using either Pacific Biosciences’ PacBio RS II (average read length exceeding 10 kb; polymerase-based) or Oxford Nanopore Technologies’ PromethION (potential read length of more than 2 Mb; synthesis-free) (Sone et al. 2019). Long-read sequencing (LRS) in particular is a burgeoning opportunity for repeat disease gene discovery and will be covered in more detail in the following section.
Another critical technological advancement that is facilitating greater ability to study repeat sequences is the production of high-throughput sequencing data without PCR amplification (Scior et al. 2011; Hommelsheim et al. 2015). PCR amplification of repeat units is especially difficult because polymerases may not proceed completely through the tract, thus generating fragmented reads of pure repeat sequences which cannot be assembled accurately with high confidence or are inappropriately assembled (e.g., as artificial inversions). Further, shorter repeats within the genome will also show PCR bias as they are more easily polymerized through than longer repeats, thereby biasing the coverage within the genome. On a per-sequence basis, artifacts and bias are also prevalent due to the high error rate of polymerases within repeat sequences, the truncated products acting as preferentially polymerized templates during subsequent rounds of polymerization, and the misalignment of primers within repetitive templates (Scior et al. 2011; Benjamini and Speed 2012; Hommelsheim et al. 2015). The development of Illumina's amplification-free sequencing technology (Kozarewa et al. 2009; Kozarewa and Turner 2011) made it possible to obtain massive SRS data with unbiased coverage of the genome. Using this technology, one of the most challenging sequences for PCR, a CGG repeat expansion, was discovered to cause NIID and related diseases (Ishiura et al. 2019), independently of the LRS-based study (Sone et al. 2019).
Perhaps the largest leap in advancement and a necessary component of identifying expanded repeats within sequencing data is the development of data analysis algorithms to correctly detect expanded TRs in a locus-specific manner from SRS data. A series of TR-expansion gene discoveries (Ishiura et al. 2018, 2019) was made with the help of TRhist (Doi et al. 2014), an algorithm specifically developed to detect and correctly annotate TR expansions. Their identification of expanded TTTCA repeats within long TTTTA repeats causing BAFME1 was also facilitated by long-read sequencing technologies. The recent discovery of GCA repeat expansions in GD (van Kuilenburg et al. 2019) was done with the help of ExpansionHunter (EH) (Dolzhenko et al. 2017), another example of software for detection of TR expansions from SRS WGS data.
In addition to TRhist and EH, there are several other algorithms for detecting TR expansions from short-read WGS data, including STRetch (Dashnow et al. 2018), GangSTR (Mousavi et al. 2019), exSTRa (Tankard et al. 2018), and TREDPARSE (Tang et al. 2017). Most of these algorithms require catalogs of all previously found repeat motifs within the reference genome. They leverage such information to detect expansions of these motifs at specified locations in the sequencing reads aligned to the reference. Initially, these algorithms were not well suited for accurate detection of TR expansions in regions with complex repeat configurations due to alignment complications and improper detection of individual repeat motifs within complex repeat tracts. Examples include: the expanded CCTG repeat causative for DM2, which is located immediately adjacent to CA and CAGA repeat tracts (Liquori et al. 2001), the expanded CAG repeat associated with Huntington disease (HD), which is followed by a CCG repeat tract (The Huntington Disease Collaborative Research Group 1993), and the complex polyalanine-coding GCN repeat which is expanded in congenital central hypoventilation syndrome (CCHS) (Amiel et al. 2003). In the face of these obstacles, a couple of improvements were made to EH. The new version (EH ver. 3.0.0) can now handle complex TR expansions with the help of catalog data from complex TR loci (Dolzhenko et al. 2019) and has been shown to be able to accurately genotype the polyalanine repeat in the CCHS-causing PHOX2B gene. EH and other catalog-based algorithms are updated to add more complex repeats to their catalogs as they are discovered; however, this requires the identification of a repeat prior to its inclusion in the catalog. As a consequence of this, these catalog-based algorithms can effectively be used to find repeats that fall within known motifs but are blind to motifs that have yet to be identified.
In the same vein, another major challenge is that some TR expansion disorders arise by “insertion” of new expanded repeat units into another repeat, such as the TGGAA repeat that appears at the TAAAA repeat locus in SCA31 (Sato et al. 2009). This interferes with correct mapping of short sequences onto the reference genome as these repeats do not exist within the reference genome. This challenge in particular is difficult to overcome because catalog-based algorithms cannot be used to identify repeat sequences that are not found within the reference genome, making these repeats invisible to these algorithms. This is complicated further by the choice of reference genome used for analysis, as shown by recent studies which determined a 1.5% and 2% discordance in SNVs and indels, respectively, between GRCh38 and the GRCh37 human reference genomes (Li et al. 2021). The human reference genome does not represent the sequence diversity of human populations. Strong examples of this shortfall include deep sequencing and contiguous assembly of the reads that did not align with the reference genome, which added 46 Mb and 296.6 Mb, respectively, of novel sequence—up to 10% of the refence human genome (Sherman et al. 2019; Eisfeldt et al. 2020). Also, a given human population cannot be represented by a single reference genome representing distinct human populations. These new sequences were found to be enriched in STRs (28%) and satellite repeats (15%) (Eisfeldt et al. 2020), suggesting that studies that depend upon the current reference genome to identify new repeats will be handicapped. Repeat tract lengths in the reference genome are likely to be shorter than a representative of population medians. As discussed by Song et al. in 2018, lengths of TRs in the human reference genome are likely underrepresented by one or two orders of magnitude, where actual tract lengths can be 10–100 times larger than the repeat size annotated in the reference assembly (Song et al. 2018).
Another example of “insertion” of new pathogenic repeats into already existing repeats of distinct sequences is the RFC1 repeats. The pathogenic repeat motif [(AAGGG)400–2000 or (ACAGG)exp] must be present homozygously to cause CANVAS, but when present heterozygously, a nondisease state arises. In contrast, the nonpathogenic motifs, even expanded are [(AAAAG)11 or (AAAAG)exp, and (AAAGG)exp]. The recessive aspect of this mutation, and the change of the repeat motif at the same locus relative to the nonaffected population, suggests that this is a highly polymorphic repeat. That the disease-causing motifs include a seemingly a limited subset of sequences suggests that this repeat sequence is at the core of CANVAS disease (Akçimen et al. 2019), as with SCA31 (Sato et al. 2009).
The reference genome is missing either the repeat and/or some of its flanking sequences for numerous repeat-expandable genes, including CANVAS, SCA31, SCA37, BAFME1, 2, 3, 4, 6, 7, and DBQD. This is likely due to the inability of the methods used to handle the repeat. For example, the XYLT1 CGG repeat and its flanking sequences could not be easily obtained by PCR amplification of the GC-rich promoter from a healthy individual (devoid of the CGG expansion) without highly specialized conditions, and the authors suspected G-quadruplex structures as the problem (Faust et al. 2014), also a likely source for its absence in the reference genome. These sequences are unstable in bacterial vectors used for the initial sequencing of the reference genome. Retrospectively, it is understandable that the reference genome was missing the repeat and its flanking sequences. In fact, this had previously been observed for FMR1, where two reports found different sequences flanking the repeat, derived from a clone from a normal X Chromosome; it was concluded that “…the sequences missing in the Kremer report (Kremer et al. 1991) are likely an artifact of the numerous cloning steps involved in preparation of the template and further underscore the instability of the region in heterologous hosts” (Fu et al. 1991).
To some degree, the hurdles noted above may be overcome through the production of “gapless” reference genomes via long-read sequencing. These efforts are being spearheaded by the Telomere-to-Telomere (T2T) Consortium (https://sites.google.com/ucsc.edu/t2tworkinggroup), which aims to fill in the numerous gaps within the reference genome by conducting complete long-read sequencing gapless assemblies of each individual chromosome. To date, the T2T Consortium has assembled and published complete sequences for several chromosomes and have preprints of assemblies of the whole genome (Jain et al. 2018a; Miga et al. 2020; Hoyt et al. 2021; Logsdon et al. 2021; Nurk et al. 2021). The new T2T-CHM13 reference includes gapless assemblies for all 22 autosomes plus Chromosome X. As expected, many of the gaps were occupied by repeat-rich sequences such as pericentromeric regions, ribosomal DNA arrays, and large segmental duplications with high sequence similarity between duplications (Bork and Copley 2001; Eichler 2001). Such efforts reveal a massive amount of genetic information that has been impenetrably cloaked by previous sequencing efforts and hence unable to be included in many biological assessments. Specifically, they now enable the unveiling of the roles that these highly polymorphic sequences might play in biology, evolution, natural variation, and disease. For example, the heterochromatic regions of Chromosomes 1, 9, and 16 have long been known to be composed of classical satellites (Gosden et al. 1975), and these were shown to be polymorphic in length by cytogenetics (Craig-Holmes and Shaw 1971). In individuals with the rare disorder, immunodeficiency, centromeric instability, and facial (ICF) syndrome, in addition to numerous clinical presentations, their chromosomes form complex multiradial associations at the classical satellites 2 and 3 at juxtacentromeric regions of Chromosomes 1, 9, and 16 (Xu et al. 1999). The satellite repeats at the heterochromatic region of Chr 9 are involved in pericentromeric inversions of Chr 9 (Gosden et al. 1981) and are thought to be linked to a variety of diseases (Mohsen-Pour et al. 2021). Length variations of satellite tracts on Chromosomes 1, 9, and 16 were thought to be associated with both the multiradials in ICF and pericentromeric inversions of Chr 9 (Gosden et al. 198l; Luciani et al. 2005). A deeper appreciation of satellite repeat tract length variations, and possibly sequence purity, gained by long-read sequencing could reveal associations of disease variation for these and other repeat-rich regions. Another huge advance is the discovery of the huge numbers of previously uncataloged repeats, definitively revealing that the repetitive content in the human genome is 53.9% in CHM13 (Hoyt et al. 2021).
To specifically address the current issue of the absence of a repeat motif in the reference genome, ExpansionHunter Denovo (EHdn) (Rafehi et al. 2019; Dolzhenko et al. 2020) was developed to roughly infer the genomic location (within ∼1 kb) and repeat size of “de novo” TR unit expansions (sequence motifs not present in the reference genome) within de novo assemblies of SRS data in a catalog-free manner. Independent of the work by Cortese et al., Rafehi et al. have used EHdn to identify expanded TRs in the WGS data of CANVAS-affected individuals (Cortese et al. 2019; Rafehi et al. 2019). They successfully found expanded AAGGG repeats in both alleles of an intron of the gene RFC1, where the reference sequence harbors (AAAAG)11. The first discovery of the RFC1 repeat expansion required considerable efforts that were time-consuming, as evidenced by Cortese et al. (2019), where the rapid independent discovery clearly demonstrates the strength and usefulness of EHdn. The group also showed that no other catalog-based algorithm was able to identify the complex repeat motif as it was not found within the reference genome.
More recently, EHdn, coupled with a novel outlier detection approach, led to the discovery of 2588 loci with TR expansions associated with ASD (Trost et al. 2020). This is the first time a heterogeneous complex disorder was linked to a variety of TR expansions. The reported loci are located in genes that were previously linked to ASD (such as FMR1), and many other genes that are responsible for nervous system development—a novel functional pathway for ASD that would otherwise have not been recognized by using other approaches. As much as 42.3% of the identified TRs in this study have not been previously reported. Even for the ones that were previously reported, 6% of them had at least one repeat sequence that was not present in the reference genome. These findings were bolstered by another recent publication which also identified TRs associated within a separate cohort of ASD individuals, using a novel bioinformatic tool called MonSTR (Mitra et al. 2021).
Given the substantial genetic overlap between neurodevelopmental disorders such as ASD and schizophrenia (Cross-Disorder Group of the Psychiatric Genomics Consortium 2013; Grove et al. 2019), it is likely that TR expansions may also be involved in other related disorders. Indeed, Mitra et al. (2021) also identified TRs which clustered near GWAS signals for schizophrenia and educational attainment within their ASD cohort, and a recent study also identified repeat expansions known to be associated with monogenic neurological diseases within a separate cohort of schizophrenia patients (Mojarad et al. 2021a). These recent studies highlight the necessity of developing tools which enable reference-free assembly and interrogation of the genome. Moreover, they highlight the need to be aware of the degree of genetic variation possible and hence be broad-minded in future developments.
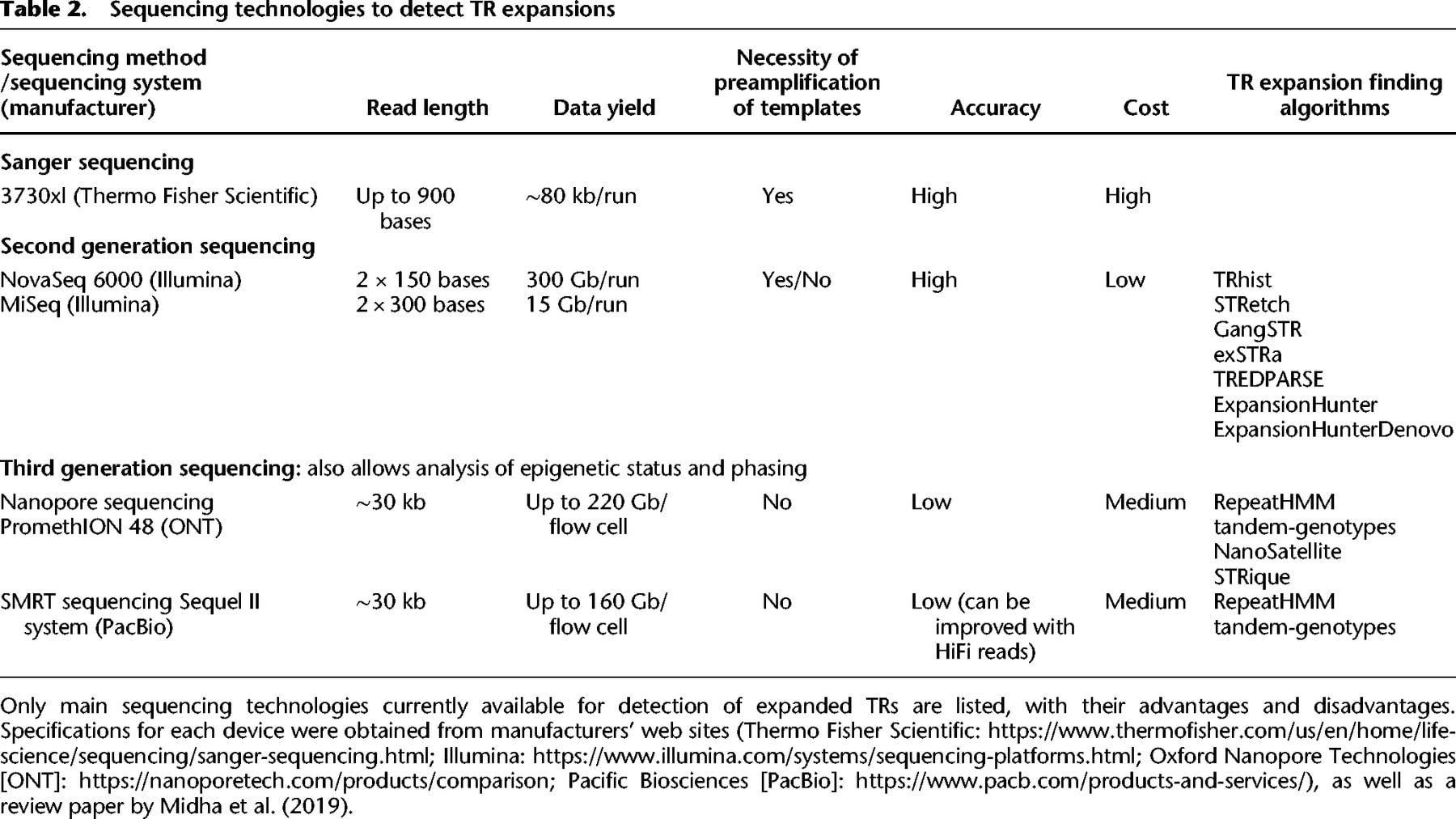
However, while these algorithms are necessary for the accurate detection of expanded repeats within SRS data, the threshold length for many of the disease-causing TRs is close to or beyond the typical short sequence read length of 100–150 bases. Algorithms like EH can infer repeat lengths from SRS data, but their accuracy is still not sufficient to make reliable diagnoses (Bahlo et al. 2018). As such, this key limitation arising from the short read length need to be complemented by LRS technologies. The advantages and disadvantages of different sequencing technologies are summarized in Table 2.
Sequencing technologies to detect TR expansions
Long-read sequencing is expected to unveil longer disease-associated TRs
The development of third generation sequencing technologies, namely Pacific Biosciences’ single molecule real-time (SMRT) sequencing (Rhoads and Au 2015) and Oxford Nanopore Technologies’ nanopore sequencing (Jain et al. 2018b), made it possible to obtain long-read data. Besides their advantages in de novo assembly, structural variant analysis, and haplotype phasing, their abilities to analyze single molecules without GC bias allowed the discoveries of disease-causing stretches of GC-rich repetitive sequences as described above (Ishiura et al. 2019). While their current cost, data generation speed, read depth, and base-calling accuracy are inferior to SRS (Midha et al. 2019), methods and analytical tools for improvements have been under development. One example is consensus circular sequencing (CSS) applied to PacBio's SMRT sequencing (Li et al. 2014). This method obtains the consensus sequence from multiple passes of a circular single molecule made by ligating both ends of the same double-strand DNA to form the circular template. While each pass produces an error-prone read, the accuracy of consensus sequences obtained by CSS has been shown to be comparable to SRS parallel sequencing in the setting of WGS (Wenger et al. 2019). Another method that is being explored to increase coverage and depth at regions of interest via Nanopore PromethION sequencing is through selective enrichment of the region—especially useful for analysis of expanded repeats which might present with repeat length mosaicism within patients. For example, Giesselmann et al. (2019) used a CRISPR-Cas12a/Cas9 approach to enrich for C9orf72-associated GGGGCC repeats and FMR1-associated CGG repeats, increasing coverage from 10 reads to nearly 100 (Cas12a) and 1000 (Cas9) reads specifically at the repeat region. The same group also combined this approach with a novel algorithm, STRique, to determine repeat length and methylation status of these repeats.
Harnessing these LRS technologies with new algorithms is rapidly becoming a more common strategy, and the data generated in this way provides a powerful tool for discovery of longer and more complex repeats. The discovery of NIID by Sone et al. (2019) was facilitated by the development of tandem-genotypes (Mitsuhashi et al. 2019), an algorithm to detect expanded TRs in long-read WGS data. Prior to this, an algorithm called RepeatHMM was published and shown to be able to accurately measure pathogenic CAG expansions in the ATXN3 gene causing Machado-Joseph disease/SCA3 and long expansions of ATTCT repeats resulting in SCA10 (Liu et al. 2017).
LRS also enables analyses of TRs with longer repeat units that are known to be associated with various complex disorders, such as Variable Number of TRs (VNTRs). VNTRs is a broad ill-defined category of TRs ranging from 6 bp to 10 kb, such as the 99-mer repeat expansion recently discovered to cause skeletal muscle disease (Ruggieri et al. 2020), or the MUC6 VNTR which has a repeat unit size of ∼507 bp and is suggested to be associated with an increased risk of Alzheimer disease (AD) (Katsumata et al. 2020; Nelson et al. 2020). Several tools have been developed to detect or genotype VTNR with short reads (Bakhtiari et al. 2018; Lu et al. 2021). However, short-read sequencing of these regions is typically difficult, often resulting in low mapping quality scores and a number of calls that fail to pass quality control filters, resulting in them being “dark and camouflaged” regions of the genome that were largely excluded from prior analysis (Nelson et al. 2020). Recent LRS studies have been instrumental in shining a spotlight on these regions. For example, a 300- to 10,000-bp VNTR in the ABCA7 gene, whose expansions are associated with increased AD risk, was recently identified from LRS data obtained by nanopore sequencing (De Roeck et al. 2019).
Recent studies highlighted the need for LRS-specific algorithms for analysis of LRS data (DeJesus-Hernandez et al. 2021; Guo et al. 2021; Miller et al. 2021). While various combinations of base-caller algorithms and tandem-genotypes could provide estimates for the ABCA7 VNTR length, expanded alleles reaching more than 10 kb were better captured by a newly developed algorithm, NanoSatellite, which directly assesses electronic current data such as that obtained by nanopore sequencing (De Roeck et al. 2019). Another example is a study made with nanopore sequencing to assess the D4Z4 macrosatellite repeat (3.3 kb/unit) number at the facioscapulohumeral muscular dystrophy 1 (FSHD1) locus (Mitsuhashi et al. 2017). Nonaffected D4Z4 alleles are polymorphic with 11–100 repeat units, whereas FSHD1-affected individuals have 10 or fewer units (Mostacciuolo et al. 2009; Pearson 2010). Mitsuhashi et al. applied LAST, a computational analysis tool developed to detect segmental duplications (Kiełbasa et al. 2011) for this purpose. Although this method still needs validation, it clearly illustrated the advantages of LRS in the analyses of longer TRs. To date, little is known about diseases caused by long TRs, and LRS is expected to open the door to this field. Moving forward, the development of LRS will likely benefit from leveraging pre-existing short-read sequencing data sets and/or integration of synthetic long reads (such as single-molecule optical maps), such that three- or two-way hybrid assembly between the different assemblies can be used to assess assembly conflicts/errors, highlight misassemblies, maximize base calling accuracy, and limit false positives (Amarasinghe et al. 2020). Even in nonrepetitive sequences, this has also shown to be a powerful approach—for example, optical maps were previously shown to greatly facilitate the resolution of three erroneously linked chromosome-scale contigs derived from SMRT-based LRS in relatives of Arabidopsis thaliana (Jiao et al. 2017). However, the strength of this approach is especially potent for repeat sequences—for example, the use of three-way integration of SRS, LRS, and optical mapping data facilitated the characterization of 36 previously unidentified large repetitive regions in the Eurasian crow, most of which were complex arrays of 14-kb satellite repeats (Weissensteiner et al. 2017). These studies clearly show the potential for LRS both as an independent approach and in tandem with pre-existing approaches.
Part 2: Technological advances complement clinical and biological understanding of repeat disease pathogenesis to facilitate repeat disease gene discovery
Various mechanisms of expanded TR toxicity
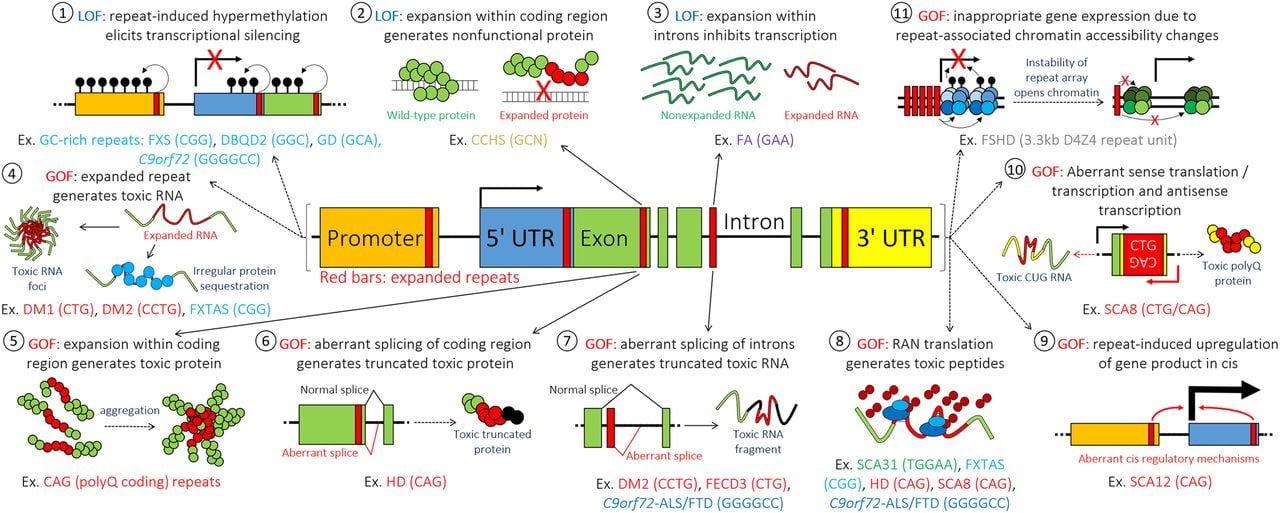
Although this current review does not focus on disease mechanisms, over the next sections we will discuss how clinical understanding, coupled with technological advances, greatly facilitates the discovery of pathogenic repeats and the development of therapeutic options. As such, here we will briefly review the mechanisms of expanded repeat toxicity. To date, at least 11 different pathogenic mechanisms have been proposed on how pathogenic TRs elicit toxicity (Fig. 2):
Loss-of-function (LOF) due to transcriptional silencing: for example, CGG expansions within the 5′ UTR of FMR1 in FXS (Pieretti et al. 1991; Devys et al. 1992; Knight et al. 1993) or the gene promoter in progressive myoclonus epilepsy type 1 (Lafrenière et al. 1997; Virtaneva et al. 1997) causes hypermethylation of the repeat and adjacent CpG islands and pathogenically silences gene transcription. While this mechanism has been known for some time, it has been understudied as a potential cause of other diseases where general loss-of-function is suspected. However, two of the latest repeat disease discoveries were shown to elicit pathogenicity through this mechanism: in Desbuquois dysplasia 2 (DBQD2) (LaCroix et al. 2019), the GGC repeat expansion within exon-1 is hypermethylated and this causes suppression of XYLT1 transcription, and in glutaminase deficiency (van Kuilenburg et al. 2019), the GCA repeat expansion results in insufficient glutaminase (GLS) mRNA transcription. These two findings remind us of the relevance of this LOF mechanism and suggest that similar findings may follow in the future.
LOF due to expansions within protein-coding genes: in CCHS, expansions of polyalanine-coding GCN repeats result in impaired function of the protein (Amiel et al. 2003).
LOF due to expansions within introns: in Friedreich ataxia (Al-Mahdawi et al. 2008), this causes pathogenic suppression of transcription through a variety of debated mechanisms which may overlap with epigenetic changes such as hypermethylation.
Gain-of-function (GOF) due to toxic RNA production which impinges on normal cellular function: in DM1, DM2, and fragile X-associated tremor/ataxia syndrome (FXTAS), repeat expansions in the 3′ UTR, 5′ UTR, and introns aberrantly generate expanded repeat RNA products, which are bound by various RNA-binding proteins, often causing toxic RNA foci. The RNA-binding proteins can also be sequestered away from their proper functions, leading to their LOF (Ranum and Cooper 2006).
Gain-of-function due to toxic proteins generated by coding expanded repeats: in the cases of polyglutamine (polyQ)-coding CAG repeat expansions such as HD and SCA3, the polyQ stretch results in protein misfolding and aggregation (Sisodia 1998). Understanding of this GOF toxic mechanism in particular has facilitated the development of therapeutics. Recently, gene silencing methods with antisense oligonucleotides (ASOs) and small interfering RNAs (siRNAs) have made considerable progress toward clinical application. One such ASO in advances stages of development is RG6042 (formally called IONIS-HTTRx; an ASO against the HTT gene developed by Ionis) which has been shown to lower mutant HTT protein levels in the cerebrospinal fluid of affected individuals and has satisfactory short-term safety profiles (Tabrizi et al. 2019). It is now entering a Phase 3 clinical trial. While gain-of-function aspects of the diseases may make approaches by ASOs/siRNAs appear attractive, caution should be paid to the contribution of other pathogenic mechanisms in the same individuals.
GOF due to aberrant splicing of coding repeats: in HD the expansion within the coding exon-1 disrupts correct splicing of intron-1, leading to its retention. This leads to the production of a toxic fragmented RNA product that contains the expanded repeat, which is then translated to a toxic truncated protein fragment (Sathasivam et al. 2013; Neueder et al. 2017, 2018; Franich et al. 2019).
GOF due to expansion promoting aberrant retention of introns near or encompassing repeat tracts: in DM2, Fuchs endothelial corneal dystrophy 3 (FECD3) (Fautsch et al. 2021), and C9orf72-associated ALS/FTD, the repeat prevents correct splicing of introns to generate a fragmented toxic RNA product. Similar to HD (mechanism 6), the fragmented RNA products may be translated into a toxic truncated protein (Sznajder et al. 2018).
GOF due to repeat-associated non-ATG/AUG-mediated translation (RAN-translation): in SCA31, FXTAS, C9orf72-associated ALS/FTD, and SCA8, expanded RNA aberrantly recruits translation machinery and produces toxic peptides without needing an ATG/AUG start site. The complexity of this pathogenic mechanism is exemplified by the multiple frames that are coded within the same repetitive sequence and the fact that both strands may undergo RAN translation—thereby generating a variety of different toxic peptides (Sato et al. 2009; Zu et al. 2011; Ishiguro et al. 2017; Glineburg et al. 2018; Krans et al. 2019).
GOF due to up-regulation of the nonmutant protein caused by a cis-mechanism of the expanded repeat: the increased expression of the PPP2R2B gene, associated with SCA12, appears to be caused by a cis-effect of the expanded CAG tract on the PPP2R2B gene products (Lin et al. 2010). This is a regulatory mechanism that likely acts on many of the repeat-containing genes in the genome.
GOF due to toxic proteins produced from transcription and translation across one strand of the expanded repeat and production of toxic RNA by transcription across the opposite strand: for example, in SCA8, the CAG repeat expansion within ATXN8 is transcribed and translated to a toxic polyglutamine protein, while transcription, but not translation, of the ATXN8 opposite strand, which has the complementary CUG expanded repeat in its 3′ UTR, produces a toxic CUG-RNA (Moseley et al. 2006).
GOF due to inappropriate expression of a gene encoded by the unstable repeat unit: for example, pathogenicity in FSHD1 results from the improper developmental expression of the double homeobox 4 (DUX4) gene, encompassed in the contracted 4q35 array of D4Z4 repeats (3.3 kb/unit). Each unit contains a DUX4 gene that is epigenetically activated upon contraction of the repeat array, with pathogenic levels of contraction resulting in FSHD1. This mechanism is likely to be more appreciated with the discovery of more clinical presentations associated with the variation of unstable arrays of large gene-sized repeat motifs. This highlights the importance of an awareness that very large repeat motifs can be unstable in a disease-relevant manner. Moreover, repeat contractions, and not just expansions, can be relevant.
Proposed mechanisms through which disease-associated repeats may exert toxicity. Multiple mechanisms may be active at a single locus. (RAN) Repeat-associated non-ATG, (UTR) untranslated region.
From this list of potential mechanisms of pathogenicity, it is clear that repeat disease pathobiology is highly complex, with shared or similar repeat sequences and often overlapping clinical presentations. It is also noteworthy that a single TR-associated disease likely has contributions from multiple pathogenic processes. For example, loss-of-function paths can exacerbate gain-of-function paths in some diseases (Schneider et al. 2020; Pal et al. 2021). Also, the surprising findings of RAN-translation and intron-retention indicates that we need to have an open mind to the diverse ways in which these mutations can express disease. Clearly, understanding the crossplay between different pathogenic repeat sequences and their functional pathogenic outcomes bolsters our ability to determine if pathologies of unknown cause result from similar repeat sequences.
Repeat expansions and fragile sites
One noteworthy association with LOF due to transcriptional silencing (pathogenic mechanism 1) is that the epigenetic changes associated with transcriptional silencing at expanded repeats often coincide with mapped fragile sites. All molecularly mapped folate-sensitive fragile sites are caused by expanded CGG repeats and associated with aberrant CpG methylation and silencing of the associated gene (FRAXA, FRAXE, FRAXF, FRA2A, FRA7A, FRA10A, FRA11A, FRA11B, FRA12A, and FRA16A). “Rare” fragile sites (∼40) are present in ≤5% of the population (with FRA16B being the most frequent rare fragile site [Felbor et al. 2003]) but can present in as few as a single individual. To this degree, recent advances toward identifying additional folate-sensitive fragile sites have included isolating epigenetic modifications of repeats (Garg et al. 2020), identification of all expandable CGG-tracts (Annear et al. 2021), and recent colocalization of these fragile sites to GC-rich repeat expansions have been found in ASD (Trost et al. 2020). However, such advances can only suggest the possible source of a fragile site; that a particular repeat is the cause of fragility must be molecularly mapped cytogenetically by FISH and genetics (Savelyeva and Brueckner 2014). Although the pathogenic link between the CGG expansions in each of these 10 folate-sensitive fragile sites and their associated partially penetrant symptoms is not yet clear, what is clear is that each shows aberrant epigenetic modifications coincident with CGG expansion regardless of disease presentation. For example, like FRAXA (FMR1), CGG expansions and aberrant methylation in AFF2, ZNF713, AFF3, and DIP2B at the fragile sites FRAXE, FRA7A, FRA2A, and FRA12A, respectively, are associated with intellectual disability, albeit in a small number of families (Winnepenninckx et al. 2007; Metsu et al. 2014a,b; Correia et al. 2015). This raises the possibility that these, and other fragile sites associations with GC-rich repeat expansions, could be more definitively linked with disease. Strengthening this hypothesis is the recent finding that the human genome contains nearly 6110 CGG repeats longer than four repeat units (found on all but the Y Chromosome)—410 of them being associated with known and candidate neurodevelopment disease genes and multiple being coincident with known fragile sites (Annear et al. 2021). Of the molecularly mapped “common” fragile sites (27 of ∼230), none are associated with a particular DNA sequence motif, repeat or otherwise (Irony-Tur Sinai and Karem 2019). It is likely that all folate-sensitive fragile sites, the largest group of rare fragile sites (∼30/∼40), are due to CGG expansions (Handt et al. 2000; Felbor et al. 2003). Other rare fragile sites, like the distamycin A-inducible rare fragile site FRA16B, is caused by an expanded AT-rich 33-bp repeat motif (Yu et al. 1997), while the bromodeoxyuridine-inducible rare fragile site FRA10B is caused by expanded AT-rich 42-bp repeat motif (Hewett et al. 1998). All fragile sites are associated with chromosomal instability (deletions, rearrangements). Both FRA10B and FRA16B have been observed homozygously in seemingly normal individuals, suggesting that at least for those individuals, at the observed ages, these expansions appear benign (Sutherland 1981; Hocking et al. 1999).
Clinical associations of specific and related repeat sequence motifs
The role of clinical geneticists cannot be underestimated. For example, as early as 1918, clinicians recognized strange transmission patterns of diseases, such as genetic anticipation in DM1 and HD (Bell 1941; for review, see Höweler et al. 1989). Similar puzzling segregation was observed for FXS (Martin and Bell. 1943). Even in the face of persuasive discrediting, based upon incorrect claims of ascertainment bias (Penrose 1947; for review, see Höweler et al. 1989), the observations of the clinical geneticist persevered and were shown to be based in the genetic instability of the repeats (Fu et al. 1991; Ashizawa et al. 1992a,b; Harper et al. 1992; Snell et al. 1993; Trottier et al. 1994). Moreover, clinicians played critical roles of both characterizing, collecting patients, and participating in genetic mapping and diagnostic aspects. These contributions are necessary ingredients to identifying disease-causing mutations. Below, we highlight recent advances of the clinical aspect in discovering repeat expansion mutations.
Some pathogenic expansions cause similar disease phenotypes according to their repeat sequence motifs, independently of the functions of genes harboring the mutations (Table 1). An early example is the discovery of CGG repeat expansion in FMR1 and the CCG/CGG repeat expansions at AFF2, ZNF713, AFF3, and DIP2B, which were all subsequently shown to be related to X-linked intellectual disability albeit in a small number of families (Knight et al. 1993; Winnepenninckx et al. 2007; Metsu et al. 2014a,b; Correia et al. 2015). Another example is the CAG/CTG repeat expansions causative for HD, SCA1, 2, 3, 6, 7, 8, 12, and 17 (Pearson et al. 2005), the discoveries of which were greatly assisted by the similarities in clinical presentation of particular motor phenotypes and pathological presentation of polyQ aggregates.
A deep clinical understanding can also facilitate understanding of underlying pathogenic mechanisms and outcomes. One such example is the similarities between DM1 and DM2 that led to the identification of the DM2 mutation. The two diseases both manifest systemic symptoms, including myotonia, muscle weakness, frontal balding, cataracts, and cardiac arrhythmias (Ricker et al. 1994). Before the identification of the DM2 mutation, researchers detected nuclear RNA foci in muscle sections of DM2-affected individuals using CUG-repeat probes against DM1 RNA foci and established that the same protein (MBNL) colocalizes with these foci (Mankodi et al. 2002). These findings led to speculation that the mutation responsible for DM2 is similar to the noncoding CTG repeat expansion that causes DM1. The DM2 mutation was subsequently revealed to be a CCTG repeat expansion in an intron of the CNBP gene (Liquori et al. 2001). The shared clinical characteristics of DM1 and DM2 are considered to be caused by toxic gain-of-function of the mutations acting in trans (Ranum and Cooper 2006). Both CUG and CCUG repeat transcripts sequester their common binding proteins, such as MBNL (Mankodi et al. 2001), and as a result, RNA metabolism is disrupted in both situations. Consistent with this model, missplicing of genes such as CLC1 (Mankodi et al. 2002) and BIN1 (Fugier et al. 2011) has been observed in both diseases.
Further, an appreciation of the clinical genetics of a disease can serve as a “red-flag” for repeat expansions—for example, the explanation of the unusual inheritance patterns of FRAXA/FXS (initially known as Martin-Bell syndrome) (Martin and Bell 1943; Sherman et al. 1984, 1985), or the explanation of genetic anticipation in DM1 or HD by repeat expansions over generations. In fact, anticipation (then called antedating) had been connected with DM1 and HD families as early as 1941 by Bell, although these findings were largely unappreciated for some time (Bell 1941). The concept was not appreciated until FXS, where repeat length directly affected the likelihood of expansion of premutations to full mutation in FXS families. This provided a mechanistic basis for the long-debated phenomenon of genetic anticipation, which in FXS is evident as incomplete penetrance accompanied by increasing likelihood of disease with subsequent generations in pedigrees (then known as the Sherman paradox). The early FRAXA studies paved the way for subsequent discoveries of repeat expansion mutations and for significant mechanistic insights.
Discoveries made in the past couple of years have strengthened the hypothesis of “repeat motif–phenotype correlation” (Ishiura et al. 2018; Ishiura and Tsuji 2020), in which toxic GOF mechanisms elicited by some expansions are the main drivers of pathogenesis, rather than altered function of the genes which contain the repeat expansion. This concept is especially useful in identifying repeat expansions which manifest similar clinical presentations. For example, in 2018, the mutation responsible for BAFME1 was identified as an insertion of an expanded TTTCA repeat into a TTTTA repeat in an intron of SAMD12 (Ishiura et al. 2018). In addition, two families manifesting indistinguishable disease phenotypes were found to have expansions of TTTCA and TTTTA repeats in introns of the TNRC6A (BAFME6) and RAPGEF2 (BAFME7) genes, respectively. As predicted by Ishiura et al. (2018), these discoveries paved the way for the identifications of other BAFME-causing repeat mutations in 2019, which have exactly the same repeat sequence in different genes: YEATS2 for BAFME4 (Yeetong et al. 2019), STARD7 for BAFME 2 (Corbett et al. 2019), and MARCHF6 for BAFME3 (Florian et al. 2019). Similar to DM1 and DM2, RNA foci of UUUCA repeats were found in autopsied brain samples of BAFME1 individuals. These observations also suggest that the trans-acting RNA gain-of-function mechanism (proposed pathogenic mechanism 4) is relevant in familial adult-onset myoclonic epilepsy. The expanded repeat causing SCA37 has the same TTTCA motif within a long stretch of TTTTA repeat (reported to be “ATTTC” repeat) (Seixas et al. 2017). BAFME patients have been reported to manifest cerebellar dysfunction (Striano et al. 2009) and atrophy (Buijink et al. 2016), and in a homozygous BAFME1 patient, histopathological findings of Purkinje cell degeneration, similar to those in SCA31 (Owada et al. 2005), have been observed. These findings connect BAFMEs with SCA37, whose cardinal clinical presentation is cerebellar ataxia and atrophy (Seixas et al. 2017). The above pieces of evidence suggest that TTTCA can now be recognized as a new common motif resulting in similar disease phenotype—another addition to the ever-growing list of diseases caused by common TR motifs.
Even more recent examples were discovered in 2019, when three groups independently found that GGC repeat expansions in an intron of NOTCH2NLC/NBPF19 genes causes NIID (Ishiura et al. 2019; Sone et al. 2019; Tian et al. 2019), a neurodegenerative disorder difficult to correctly diagnose based on clinical presentations alone (Sone et al. 2016; Okubo et al. 2019). Further reports showed that an intronic CGG repeat expansion in LOC642361/NUTM2B-AS leads to oculopharyngeal myopathy with leukoencephalopathy (OPML), and the same repeat expansion in LRP12 is responsible for oculopharyngodistal myopathy (OPDM1) (Ishiura et al. 2019). These findings were truly eye-opening for neurologists and neuropathologists, as they provided critical insight into the fundamental pathogenesis of these diseases and connected degenerative disorders of the central and peripheral nervous systems and muscles. NIID is a disease that mainly affects the brain and the peripheral and autonomic nervous systems, manifesting a variety of symptoms including dementia, tremor, cerebellar ataxia, and autonomic failure (Sone et al. 2016). On the other hand, OPDM is a type of muscular dystrophy that causes facial, bulbar, and distal weakness (Satoyoshi and Kinoshita 1977). Now, with the identification of GGC repeats as the cause of both NIID and OPDM diseases, and the knowledge that there are overlapping symptoms and radiological findings among them (Ishiura et al. 2019), we have a view that there is a clinical spectrum with NIID on one end, OPDM on the other end, and OPML in the middle. There are also strong clinical similarities between NIID and FXTAS, the classic example of neurodegenerative disease caused by CGG repeat expansion (Hagerman et al. 2001). The disease spectrum suggested by these new findings is a completely new concept for physicians, pathologists, and geneticists, one that could not have been imaginable without the discovery of its genetic cause.
If we are to accept the hypothesis that particular repeat unit sequence expansions are pathogenic and cause diseases independently of their genomic locations, it is essential to establish the possibility that these de novo TR unit expansions may also be associated with similar diseases that have yet to be identified. In the past, prototypical approaches were taken to search for trinucleotide repeat expansions in a locus-independent manner simply by looking for expansions of known disease-causing motifs (RED assay and its variations and catalog-based algorithms more recently, as described above). Now that we are equipped with high-throughput LRS technologies and computational analysis tools, we can search for TR expansions from NGS data. New discoveries are expected to follow, which will further broaden our understanding of the biological roles of TRs and their contribution to disease.
Assessing the association of repeat expansions with disease must be conducted without bias or assumptions
Several recently reported repeat disease associations occurred in unstable repeats previously thought to be clinically unimportant. For example, one of the most prevalent (affecting ∼4% of people aged 40 or over in the United States) TR expansion diseases, FECD3, was initially missed because of a clinical bias toward the expectation that the disease would present with neurodegeneration. Due to the absence of neurodegenerative phenotypes, the initial discoveries of the CTG18.1 repeat expansion (the genetic cause of FECD3) in 1993 (Schalling et al. 1993) and 1997 (Breschel et al. 1997) led to an incorrect assumption that the expansion was benign. It was not until 20 years later that the CTG repeat expansions within the TCF4 gene (CTG18.1) were linked with FECD3 (Wieben et al. 2012; Fautsch et al. 2021). In another case, an expanded CGG repeat reported at FRA16A, which in the heterozygous state has no obvious clinical impact (Nancarrow et al. 1994), was subsequently found to result in autosomal recessive skeletal disorder DBQD2 when CGG expansions were present in a homozygous state (LaCroix et al. 2019). Because the initial identifications of pathogenic TR expansions were mostly of dominant or X-linked diseases (with the exception of Friedreich ataxia), the possibility of recessive diseases tended to be overlooked, but the recent discoveries demonstrate that this is a groundless assumption (Cortese et al. 2019; LaCroix et al. 2019; van Kuilenburg et al. 2019; Pagnamenta et al. 2021).
Similarly, effects of TR length on varied clinical presentations must also be considered. One example is the human androgen receptor (AR), where the function of the gene product is well characterized, unlike the genes for most TR expansions. The AR gene contains an exonic polymorphic CAG repeat in which 95% of individuals inherit between 16 to 29 CAG repeats, encoding a polyglutamine tract. Nonrepeat LOF mutations of AR, and hence an absence of androgen receptor activity, lead to masculine feminization (androgen insensitivity syndrome; AIS), a non-neurological presentation. In contrast, expansions of the CAG tract (average size of ∼47 units in patients) lead to spinal bulbar muscular atrophy (SBMA). While not evident in 1991, the contrasting phenotypes of the LOF mutations with the CAG expansions provided support for the pursuit of a GOF toxicity path. Albeit that in most neurologically affected SBMA individuals, the AR is still functional, they do show signs of AIS (La Spada et al. 1991). Biochemically, the length of this encoded polyglutamine tract inversely affects AR transcriptional activity (Mhatre et al. 1993; Chamberlain et al. 1994; Kazemi-Esfarjani et al. 1995). Very large expansions (68 or 72 repeats) lead to severe clinical presentations of both SBMA and AIS consistent with reduced transactivation activity with long CAG tracts (Mhatre et al. 1993; Kazemi-Esfarjani et al. 1995; Grunseich et al. 2014; Madeira et al. 2018). Within the normal range, longer AR CAG tracts (>∼21 repeats) have been associated with male infertility, breast cancer, osteoporosis, and male-to-female transsexualism (Summers and Crespi 2008; Hare et al. 2009), while shorter tracts in the normal range have been associated with prostate cancer, head and neck cancer, colorectal cancer, cardiac disease, and cognition and behavior disorders (Summers and Crespi 2008). In cancers, somatic variation of the CAG repeat was biased to contracted repeats (Ferro et al. 2002; Di Fabio et al. 2009), presumably due to the increased androgen sensitivity of the AR protein, with shorter tracts giving those cells a growth advantage (Mhatre et al. 1993; Kazemi-Esfarjani et al. 1995). Other studies suggest that gender incongruence/dysphoria in the transgender woman (male-to-female) population have significantly longer polymorphic CAG repeat sequences in the AR gene, which may affect antenatal androgen activity and possibly contribute to gender incongruence (D'Andrea et al. 2020). Thus, one gene, depending upon mutation type and/or TR tract length can display extremely variable clinical manifestations and demonstrates how understanding of the natural function of the gene can serve as a guide to elucidating its mechanism of disease action. For FMR1, nonrepeat-related null LOF mutations were identified following the discovery of the fragile X-associated expansion. In the case of FMR1, intragenic deletions, nonsense changes (Wöhrle et al. 1992; Hirst et al. 1995; Lugenbeel et al. 1995), and a missense mutation, I304N (De Boulle et al. 1993) led to severe fragile X syndrome, confirming the LOF associated with the CGG expansion. Similarly, increases in FMR1 copy numbers can also lead to similar phenotypes (Rio et al. 2010). New sequencing and informatic analyses should facilitate such future pathogenic connections.
Another crucial point that must be highlighted is the delayed recognition of distinct clinical presentations and diseases associated with different expansion lengths within the FMR1 gene. Full expansion of the CGG repeat (>200 CGG) in FMR1, coupled with aberrant DNA CpG methylation, is widely known to cause the intellectual disability syndrome FXS, a phenotype recognized since 1943 (Martin and Bell 1943). It wasn't until 58 years after the description of the first FXS pedigree that individuals with unmethylated FMR1 expansions of 50–200 CGG units were distinctly characterized within a separate disease, which presented with late onset tremor and ataxia, now known as FXTAS (Hagerman et al. 2001). Prior to this, these individuals were referred to as “premutation” or “normal transmitting males/females,” a term highlighting their limited FXS phenotypes and their ability to pass on the FXS-eliciting expansion to their children, despite their distinct clinical presentations of ataxia later in life (Loesch et al. 1994). Although ataxia had previously been observed in families with Martin-Bell syndrome/FXS, prior to the discovery of the disease-causing CGG expansions (Howard-Peebles 1980), because the ataxia and intellectual disability did not simultaneously appear in a single individual, Dr. Howard-Peebles stated “Several family members are ataxic…[which] appears to be an unusual variety of spinocerebellar atrophy… [and] There appears to be no relationship between this disorder and X-linked mental retardation with a fragile Xq”. These early observations of FXTAS were also complicated by limited family member numbers and the multiplicity of phenotypes resulting from different expansion lengths, which has only been delineated within the early 2000s (Jacquemont et al. 2004; Rodriguez-Revenga et al. 2009). This penetrance issue also complicated early characterization of DM1 and other diseases (Echenne and Bassez 2013; De Antonia et al. 2016; Joosten et al. 2020). Despite this, it is highly laudable that Dr. Howard-Peebles noted the ataxia in the FXS family, as it facilitated the eventual correct characterization of the ataxia as a separate disorder (Howard-Peebles 1980). While it is unlikely that this is the first time such an association was clinically observed, it may be the first published description of FXTAS in FXS families. Retrospectively, it is understandable how an association between FXS and the late-onset, slowly progressive motor symptoms of FXTAS had been overlooked for such a long time, but this should serve as a teaching lesson: ascertainment bias by human involvement may lead to missed genetic attributions of varied clinical presentations (symptoms, ages at onset, etc.) to a single TR locus. For example, a confounding factor in the case of FXS is that families (boys with FXS and their mothers) were typically under the care of pediatricians, while the grandfathers at risk for FXTAS were seen by separate specialists (neurologists, geriatricians).
A similar example occurred in cases of nonneuropsychiatric clinical presentations in FXS family members, which were met with delayed recognition and acceptance by the research community for being genetically linked to CGG expansions in FMR1. In the late 1980s to early 1990s, studies of FXS families suggested that non-FXS premutation mothers were at risk of early menopause and increased rates of dizygotic twinning, which in 1995 led to testing an association of ovarian failure in these women (Conway et al. 1995; Murray et al. 1998; Sherman 2000). A link between premutation (CGG) 55–199 lengths in FMR1 and fragile-X associated primary ovarian insufficiency (FXPOI) is now accepted, and recently a study of 1668 women has refined the risk of FXPOI: specifically, females with 85–89 repeats are at the highest risk, while those with 55–65 repeats or 120–199 repeats did not have a significantly increased risk for FXPOI compared to women without any CGG expansions <45 repeats (Allen et al. 2021). The risk of early menopause was very similar (Allen et al. 2021). A link of twinning rates and FMR1 premutations remains an enigma with inconsistent claims (for and against an association), likely due to studies that do not account for repeat size and possibly timing of twinning relative to X-inactivation (Sherman 2000; Allen et al. 2007). Thus, very distinct clinical presentations can arise from repeat expansions in a given gene, and these can critically depend upon repeat expansion size.
With the clarity of hindsight, it is easy to overlook that late-onset genetic disorders, especially those that show incomplete penetrance, are challenging to study through families—especially so in the era prior to molecular biology. A thought experiment is instructive here: without the link provided by individuals ascertained through fragile X syndrome, would it have been possible to define the partially-penetrant FXTAS or FXPOI? It seems unlikely, and this lesson should be carried with researchers into the future, especially with the expansion of molecular biology research, as similar breadths of clinically diverse presentations may also be linked to expansion size at the DM1 (Trost et al. 2020), C9orf72 (Miller et al. 2016; Van Mossevelde et al. 2017; Fredi et al. 2019; Tábuas-Pereira et al. 2019), the NIID loci (Sone et al. 2016), FMR1 (Schneider et al. 2020), or any other of the known or yet undiscovered repeat diseases. To this degree, clinicians and researchers must share observations, be open-minded and embrace the likelihood that repeat diseases are not limited to only continuums of severity of one but of potentially diverse phenotypes.
The length of repeats in one gene can predispose to distinct diseases
Recent studies have started to unveil how the nondiseased size of a TR in one disease-associated gene can be linked to the susceptibility of another distinct disease. For example, the intermediate CAG repeat lengths of ATXN2 gene has been identified as a risk factor for developing ALS (Elden et al. 2010; Conforti et al. 2012), SCA3 (Tezenas du Montcel et al. 2014), FTD (Fournier et al. 2018), and AD (Rosas et al. 2020). This association was further confirmed by a series of studies, and a meta-analysis of these data showed that an intermediate CAG repeat (30–33) allele in ATXN2 is associated with increased risk of developing ALS with the odds ratio of 4.44 (Wang et al. 2014).
Other instances of this phenomena are observed in the intermediate CAG repeat lengths of: ATXN1 as a risk factor for FTD, AD, ALS, SCA3, and SCA6; ATXN3 as a risk factor for SCA6 and SCA7; ATXN7 as a risk factor for SCA2; TBP as a risk factor for SCA7; and HTT as a risk factor for FTD, AD, and SCA3 (Conforti et al. 2012; Tezenas du Montcel et al. 2014; Rosas et al. 2020). The nonpathogenic HTT CAG tract length has also recently been associated with variable changes in risk for ASD, where longer allele lengths are associated with an enhanced ASD risk (Piras et al. 2020). Another observation is that the GT repeat length in the promoter region of the HMOX1 gene modulated the risk of human immune deficiency virus (HIV)-related central nervous system inflammation, such that shorter GT repeats are related with decreased risk of HIV encephalitis (Gill et al. 2018) and HIV-associated neurocognitive impairment (Garza et al. 2020). Increasing the complexity of these associations is the fact that in certain instances longer repeat lengths could also be protective—for example, with longer CAG lengths in HTT (within the normal range) being protective for SCA3 age of onset (Tezenas du Montcel et al. 2014).
The effects of TRs resulting in variable disease susceptibility can partially be explained by their effects on gene expression. A genome-wide search utilizing RNA-seq data of lymphoblastoid cell lines and lobSTR (Gymrek et al. 2012)—another software to analyze TR length—revealed 2060 STRs in association with gene expression (the authors coined these STRs as eSTRs) (Gymrek et al. 2016). This study further identified that 12 eSTRs are significantly associated with clinical phenotypes, including Crohn's disease, rheumatoid arthritis, and type 1 diabetes mellitus. The findings of eSTRs are supported by a study by Quilez et al., in which they genotyped 4849 promoter-associated STRs in 120 individuals and found more than 100 STRs associated with DNA methylation and neighboring gene expression (Quilez et al. 2016). These TRs were shown to have tendencies toward overlapping with transcription factor binding sites, providing an explanation for possible biological mechanisms of action. The same group used another STR analysis software, HipSTR, to link hundreds of eSTRs with complex disorders such as schizophrenia and inflammatory bowel disease, and complex traits including height and intelligence (Fotsing et al. 2019). A more recently developed powerful bioinformatic tool, adVNTR-NN, used a neural network to rapidly genotype 10,264 VNTRs in 652 individuals (Bakhtiari et al. 2021). Greatly improving processing times from previous tools, adVNTR-NN can genotype a single VNTR from 55× whole-genome data in 18 sec with high accuracy. The group found 163 VNTRs associated with regulation of proximal gene expression (designated eVNTRs) in 46 different tissues—with about 50% of these having a likely causal impact on the expression of proximal genes. Within the eVNTRs, several were associated with Alzheimer disease, obesity, and familial cancers, supporting that repeat-associated expression dysregulation is likely a contributing factor to pathogenesis.
Analysis of these secondary repeat instability effects offers key insights into disease and potentially illuminates therapeutic potential. For example, a recent study demonstrated that cancer cells with microsatellite instability arising from DNA mismatch repair deficiency incur previously unknown large-scale expansions of TA repeats (Van Wietmarschen et al. 2020). TA repeats are found genome-wide and, when expanded, they stalled replication forks, activated DNA damage response kinases, and required WRN helicase for processing. In the absence of WRN, however, expanded repeats were susceptible to cleavage by the MUS81 nuclease, leading to massive chromosome shattering and synthetic lethality in cancer cells. Nearly 15% of colorectal cancers, 20%–30% of endometrial cancers, 15% of gastric cancers, and 12% of ovarian cancers are caused by deficiency in DNA mismatch repair, supporting the development of therapeutic agents that target WRN for microsatellite instability-associated cancers.
Further studies are expected to clarify the role of TRs in complex human traits and diseases, which has been proposed to explain “missing heritability” (Hannan 2010) as demonstrated in the recent study on ASD (Trost et al. 2020).
Repeat tract purity and gene variance can be an issue for clinical awareness and research
Another major aspect of repeat disease genetics that is highly relevant to clinical awareness and understanding of pathogenesis is the concept of repeat tract purity—the presence or absence of nonrepeat units within a tract of tandemly repeating motifs. Recently, it has become increasingly apparent that the presence of interruptions within expanded TR tracts affects genetic instability as well as age-of-disease-onset severity. Soon after the discovery of repeat expansions as a cause of disease, it was found that interruptions within the repeat tract stabilized these repeats against expansions, whereas loss of interruptions makes the repeat susceptible to expansion (Chung et al. 1993). Interrupted nonexpanded repeat tracts are typically associated with beneficial aspects for FXS, SCA1, SCA2, and HD (Eichler et al. 1994; Chong et al. 1995; Latham et al. 2014)—protecting against germline and somatic repeat instability and, in this manner, “protecting” against disease aspects for the gene in which the repeat resides. The purity of the FMR1 premutation CGG tracts, when diagnostically assessing the AGG interruptions in the premutation CGG expansions in FMR1 by single-molecule PacBio sequencing, allows accurate risk estimates for having a child with FXS (Ardui et al. 2018). Sequencing has many advantages over PCR-based methods and provides improved genetic counseling for women with a premutation—for example, in decisions of family planning.
Because the size of the expansion correlates with disease severity, inhibition of repeat expansions was hypothesized to drastically modulate age-of-disease-onset severity. Indeed, recent data supports this hypothesis by revealing that, in a portion of individuals with HD (Ciosi et al. 2019; Genetic Modifiers of Huntington Disease (GeM-HD) Consortium 2019; Wright et al. 2019), the absence of CAG tract purity may have strong effects on the age-of-onset, disease progression, severity, and phenotypic manifestations. For example, in HD, the polyQ-coding CAG repeat usually ends with CAACAG (which also codes for QQ) or (CAACAG)2 (which codes for QQQQ), but in some HD individuals, the interrupting CAA units are absent. The groups found that those carrying the (CAACAG)2 interruption had significantly delayed disease age-of-onset and lessened severity, and those carrying no interruption had significantly hastened age-of-onset and worsened severity, relative to those carrying a single CAACAG—despite all expressing a mutant HTT protein with the same polyQ length. These facts may suggest that pure repeat tracts are more susceptible to somatic repeat instability and thus result in earlier disease onset and more severe phenotypes. As such, correct identification of interrupted repeats within patient cohorts is essential for planning clinical trials, providing prognostic insight, and in conducting patient research.
It should also be noted that interrupted repeats can also have deleterious attributes. As mentioned in the previous section, larger nonpathogenic length repeats can affect presentation of other diseases—for example, larger ATXN1 or AXTN2 CAG tracts within the wild-type range can be associated with ALS, FTD, AD, and SCA3. These larger tract sizes are typically interrupted CAG tracts (Corrado et al. 2011; Yu et al. 2011; Conforti et al. 2012). While the manner by which the interruptions contribute to disease predisposition is unknown, a broader appreciation of repeat purity is clearly wanting.
While the presence of repeat tract interruptions on shorter/nonexpanded tracts has long been known (Eichler et al. 1994; Latham et al. 2014), the assessment of purity of longer/expanded tracts and complex motifs is more challenging. For example, while long-presumed to be pure, long disease-associated expansions of the myotonic dystrophy CTG tract have been shown to be interrupted with various non-CTG units, with unusual patterns. These interrupted alleles have been associated with altered predispositions to germline and somatic instabilities and may be associated with vastly altered clinical presentations (Musova et al. 2009; Braida et al. 2010; Santoro et al. 2013, 2017; Botta et al. 2017; Cumming et al. 2018; Tomé et al. 2018; Ballester-Lopez et al. 2020). While LRS is expected to reveal the inaccessible areas of long stretches of TRs, the high error rates of nanopore sequencing and SMRT sequencing technologies appear as obstacles to fine analysis and may introduce “artificial interruptions.” While CSS has been applied to some expanded TRs, such as GGGGCC repeat expansion in C9orf72 (Ebbert et al. 2018) and CGG repeat expansion in NOTCH2NLC (Sone et al. 2019), its usefulness to characterize the purity of TRs still needs more validation. Currently, interruptions still present a major challenge for LRS that will need to be addressed as the field progresses. A full appreciation of the purity of any expanded repeat will likely lead to improvement to clinical, diagnostic, and genetic counselling. The evolution of bioinformatic tools is wanting.
In addition to repeat tract purity, another major modifier of disease is naturally occurring variants of genes which act as trans-modifiers of disease. Of note are the DNA repair gene variants known to modify disease presentation in patients, likely by modifying the level of somatic expansions at the repeat. For example, recent age-of-onset for HD GWASs have identified SNP variants in the DNA repair genes MSH3, FAN1, PMS2, LIG1, and MLH1 (Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium 2019), and a separate GWAS identified variants of MSH3 modified somatic instability and disease severity in HD and DM1 patients (Flower et al. 2019). Corroborating these findings and illuminating overlap between the different diseases, a separate GWAS identified FAN1 and PMS2 variants as significant modifiers of age-of-onset for several different CAG expansion SCAs (Bettencourt et al. 2016). These studies demonstrate the importance of these DNA repair proteins in disease pathogenesis. Furthermore, their impact is not limited to CAG/CTG disorders, as a recent GWAS in XDP (caused by a CCCTCT repeat expansion) also identified variants of MSH3 and PMS2 as significant modifiers of age-of-onset (Laabs et al. 2021). While these SNP-based approaches sifting known variants illuminate novel shared pathways that may contribute to pathogenicity, large-scale whole-genome sequencing efforts are likely to reveal novel gene variants that are significant modifiers of disease (e.g., see Deshmukh et al. 2021).
Beyond humans and beyond disease
Nonhuman organisms can display TR length variations with associated disease or biological consequences. Naturally occurring repeat length variations are implicated in disease of nonhuman organisms (summarized in Table 3) and in human nondisease phenotypes, such as height. Prominent examples include various canine diseases associated with repeat expansions, such as (1) the dodecamer GCCGCCCCCCGC pathogenic repeat associated with a epilepsy (canine Lafora disease) in many species of dogs (Lohi et al. 2005; Webb et al. 2009; Barrientos et al. 2019; Kehl et al. 2019; for review, see von Klopmann et al. 2021), (2) a 38-bp VNTR in the dopamine transporter gene, DAT/SLC6A3, associated with seizures and behavioral issues in Belgian Malinois dogs (Lit et al. 2013), and (3) the GAA repeat expansion associated with spinocerebellar ataxia in Italian Spinone dogs (Forman et al. 2015). Oddly, nonrepeat mutations in the human homologs of some of these genes, like NHLRC1 and ITPR1, cause similar disease in humans, but the human gene does not contain the unstable repeat present in the canine gene as outlined in Table 3 (Chan et al. 2003; Das et al. 2017; Zambonin et al. 2017; for review, see von Klopmann et al. 2021). While it remains puzzling that the highly unstable, pathogenic “dynamic” repeat mutations seem to be mostly confined to humans, in these particular cases, it seems that the canine disease, but not the human, is linked to repeat expansions. However, there seems to be some overlap between dogs and human, albeit controversial, with VNTR variation in DAT/SCLR and behavioral presentations (Hauser et al. 2002; Lafuente et al. 2007; Ivashchenko et al. 2015). Repeat expansions, with biological consequences, have been documented in plants. For example, an expanded TTC/GAA intronic repeat within the ILL1 gene of Arabidopsis thaliana is responsible for growth defects and temperature sensitivity within a strain of the plant species (Sureshkumar et al. 2009; Tabib et al. 2016). An awareness of repeat biology in crops is only beginning—for example, different numbers of serine-encoding TCG repeats of ERF17 may regulate apple peel degreening during ripening (Han et al. 2018). In nonvertebrates, variations in repeat length within coding sequences have been suggested as a source of speciation in honey bees (Zhao et al. 2018), as a mediator of lifespan in yeast (Barré et al. 2020), and as a regulator of immunity in pearl oysters (Cao et al. 2021). Analysis of Arabidopsis thaliana reveals a large degree of genetic variability associated with natural polymorphic variants within repeat tracts in the genome; with 95% of the 2046 STR loci tested displaying significant polymorphism (Press et al. 2018). These examples exhibit that many nonhuman disease or complex phenotypes are associated with repeat length variations and could contribute to evolutionary-selectable traits which could be beneficial or detrimental.

As our understanding of repeat sequences grows, so too will our appreciation for natural variability of complex traits in humans due to variations in repeat length. For example, a recent study analyzing the genomes of 3622 Icelanders by LRS identified a median of 22,636 structural variants per person, representing 13,353 insertions and 9474 deletions spanning a total of 10 Mb per haploid genome. While some of these variations are disease-relevant (such as the 69-mer variation within NACA, associated with atrial fibrillation), some variants were associated with nondisease complex traits, such as the 57-bp repeat within ACAN which was associated with height of the individual (Beyter et al. 2021). This is coincident with previous reports which also found associations of repeat lengths with height (Fotsing et al. 2019). Limb and skull morphological variations in dogs have already been linked with differences in repeat sizes of a variety of genes, suggesting that natural human variation could also be attributed to repeat length variations (Fondon and Garner 2004). Indeed, a recent large scale repeat length polymorphism analysis of 118 coding VNTRs in more than 400,000 individuals reveals associations of repeat lengths with nearly 800 different human trait phenotypes, including height, male pattern baldness, and hair morphology, and potentially disease-associated phenotypes such as lipoprotein concentration and kidney function (Mukamel et al. 2021). Thus, DNA repeat length variations may affect various phenotypes, not necessarily disease attributes only, thereby precipitating rapid phenotypic variations which may affect rates of natural section. On an evolutionary scale, TR variations that may have null or deleterious effects could, with environmental change, become advantageous.
Concluding remarks—toward future discoveries
As of December 2021, there were 63 disease-associated or disease-causing unstable TR loci, at least 22 repeat motifs (not counting complex, large, and/or variable motifs), associated with >69 diseases, where some diseases are common to some of the same TR loci. Our quest for TR expansions and their association with disease is still far from complete; currently we only see the tip of the iceberg. Recent technological progress has facilitated the unveiling of TR expansions with large effect size on clinical phenotypes, but our knowledge of those with small effect size is extremely limited. The mechanism of TR expansion has been eagerly sought after, and earlier studies indicate the impact of DNA repair proteins and their naturally occurring variants (Tomé et al. 2009). This view is supported by the recent large-scale screens for disease modifiers (Moss et al. 2017; Flower et al. 2019; Genetic Modifiers of Huntington Disease (GeM-HD) Consortium 2019), which may lead to the development of disease-modifying therapies. The impact of somatic instability of TRs is now recognized for numerous neurodegenerative disorders and cancers, but noninvasive methods to evaluate its degree in various organs and tissues are still lacking. We must pause to consider how many TR expansions may yet prove to be associated with biological functions, diseases, and evolutionary change. Further identification and understanding of TRs, beyond the tip of the iceberg, will reveal a new landscape of biology and medicine.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the Canadian Institutes of Health Research (FRN175329, R.K.C.Y.; and FRN148910, C.E.P.), the Natural Sciences and Engineering Research Council (RGPIN-2016-08355, C.E.P.), the Marigold Foundation (C.E.P.), the Petroff Family Fund (C.E.P.), and Brain Canada (R.K.C.Y.). C.E.P. holds a Tier 1 Canada Research Chair in Disease-Associated Genome Instability. The authors thank the reviewers, who provided significant, fruitful, and constructive feedback for this work. Dedication: This review is dedicated to two pioneers and scientific catalysts: Professor Sir Peter S. Harper (1939–2021), an exceptional clinical geneticist, advocate, and mentor to many in the arenas of myotonic dystrophy, Huntington disease and beyond: and Professor Stephen T. Warren (1953–2021), a pioneering human molecular geneticist, ever committed to advancing the understanding of fragile X and related disorders, a leader, advocate, and mentor to many.
Note added in proof
During the proofing stage of this review, an additional preprint has revealed an association for expanded repeats and schizophrenia risk (Mojarad et al. 2021b).
Notes
[1] Article published online before print. Article and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.269530.120.
References
- Akarsu A. 1996. Genomic structure of HOXD13 gene: a nine polyalanine duplication causes synpolydactyly in two unrelated families. Hum Mol Genet 5: 945–952. 10.1093/hmg/5.7.945
- ↵Akçimen F, Ross JP, Bourassa CV, Liao C, Rochefort D, Gama MTD, Dicaire MJ, Barsottini OG, Brais B, Pedroso JL, 2019. Investigation of the RFC1 repeat expansion in a Canadian and a Brazilian ataxia cohort: identification of novel conformations. Front Genet 10: 1219. 10.3389/fgene.2019.01219
- ↵Allen EG, Sullivan AK, Marcus M, Small C, Dominguez C, Epstein MP, Charen K, He W, Taylor KC, Sherman SL. 2007. Examination of reproductive aging milestones among women who carry the FMR1 premutation. Hum Reprod 22: 2142–2152. 10.1093/humrep/dem148
- ↵Allen EG, Charen K, Hipp HS, Shubeck L, Amin A, He W, Nolin SL, Glicksman A, Tortora N, McKinnon B, 2021. Refining the risk for fragile X-associated primary ovarian insufficiency (FXPOI) by FMR1 CGG repeat size. Genet Med 23: 1648–1655. 10.1038/s41436-021-01177-y
- ↵Al-Mahdawi S, Pinto RM, Ismail O, Varshney D, Lymperi S, Sandi C, Trabzuni D, Pook M. 2008. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum Mol Genet 17: 735–746. 10.1093/hmg/ddm346
- ↵Amarasinghe SL, Su S, Dong X, Zappia L, Ritchie ME, Gouil Q. 2020. Opportunities and challenges in long-read sequencing data analysis. Genome Biol 21: 30. 10.1186/s13059-020-1935-5
- ↵Amiel J, Laudier B, Attié-Bitach T, Trang H, de Pontual L, Gener B, Trochet D, Etchevers H, Ray P, Simonneau M, 2003. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet 33: 459–461. 10.1038/ng1130
- ↵Annear DJ, Vandeweyer G, Elinck E, Sanchis-Juan A, French CE, Raymond L, Kooy RF. 2021. Abundancy of polymorphic CGG repeats in the human genome suggest a broad involvement in neurological disease. Sci Rep 11: 2515. 10.1038/s41598-021-82050-5
- Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan X-Y, Sauter G, Kallioniemi O-P, Trent JM, Meltzer PS. 1997. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277: 965–968. 10.1126/science.277.5328.965
- ↵Ardui S, Race V, de Ravel T, Van Esch H, Devriendt K, Matthijs G, Vermeesch JR. 2018. Detecting AGG interruptions in females with a FMR1 premutation by long-read single-molecule sequencing: a 1 year clinical experience. Front Genet 9: 150. 10.3389/fgene.2018.00150
- ↵Ashizawa T, Dubel JR, Dunne PW, Dunne CJ, Fu Y-H, Pizzuti A, Caskey CT, Boerwinkle E, Perryman MB, Epstein HF, 1992a. Anticipation in myotonic dystrophy: II. Complex relationships between clinical findings and structure of the GCT repeat. Neurology 42: 1877–1883. 10.1212/WNL.42.10.1877
- ↵Ashizawa T, Dunne CJ, Dubel JR, Perryman MB, Epstein HF, Boerwinkle E, Hejtmancik JF. 1992b. Anticipation in myotonic dystrophy: I. Statistical verification based on clinical and haplotype findings. Neurology 42: 1871–1877. 10.1212/WNL.42.10.1871
- ↵Aslanidis C, Jansen G, Amemiya C, Shutler G, Mahadevan M, Tsilfidis C, Chen C, Alleman J, Wormskamp NGM, Vooijs M, 1992. Cloning of the essential myotonic dystrophy region and mapping of the putative defect. Nature 355: 548–551. 10.1038/355548a0
- ↵Bahlo M, Bennett MF, Degorski P, Tankard RM, Delatycki MB, Lockhart PJ. 2018. Recent advances in the detection of repeat expansions with short-read next-generation sequencing. F1000Res 7: 736. 10.12688/f1000research.13980.1
- ↵Bakhtiari M, Shleizer-Burko S, Gymrek M, Bansal V, Bafna V. 2018. Targeted genotyping of variable number tandem repeats with adVNTR. Genome Res 28: 1709–1719. 10.1101/gr.235119.118
- ↵Bakhtiari M, Park J, Ding Y-C, Shleizer-Burko S, Neuhausen SL, Halldórsson BV, Stefánsson K, Gymrek M, Bafna V. 2021. Variable number tandem repeats mediate the expression of proximal genes. Nat Commun 12: 2075. 10.1038/s41467-021-22206-z
- ↵Ballester-Lopez A, Koehorst E, Almendrote M, Martínez-Piñeiro A, Lucente G, Linares-Pardo I, Núñez-Manchón J, Guanyabens N, Cano A, Lucia A, 2020. A DM1 family with interruptions associated with atypical symptoms and late onset but not with a milder phenotype. Hum Mutat 41: 420–431. 10.1002/humu.23932
- ↵Bamshad MJ, Nickerson DA, Chong JX. 2019. Mendelian gene discovery: fast and furious with no end in sight. Am J Hum Genet 105: 448–455. 10.1016/j.ajhg.2019.07.011
- ↵Barré BP, Hallin J, Yue J-X, Persson K, Mikhalev E, Irizar A, Holt S, Thompson D, Molin M, Warringer J, 2020. Intragenic repeat expansion in the cell wall protein gene HPF1 controls yeast chronological aging. Genome Res 30: 697–710. 10.1101/gr.253351.119
- ↵Barrientos L, Maiolini A, Häni A, Jagannathan V, Leeb T. 2019. NHLRC1 dodecamer repeat expansion demonstrated by whole genome sequencing in a Chihuahua with Lafora disease. Anim Genet 50: 118–119. 10.1111/age.12756
- ↵Bell J. 1941. On the age of onset and age at death in hereditary muscular dystrophy with some observations bearing on the question of antedating. Ann Eugen 11: 272–289. 10.1111/j.1469-1809.1941.tb02290.x
- ↵Bell MV, Hirst MC, Nakahori Y, MacKinnon RN, Roche A, Flint TJ, Jacobs PA, Tommerup N, Tranebjaerg L, Froster-Iskenius U, 1991. Physical mapping across the fragile X: hypermethylation and clinical expression of the fragile X syndrome. Cell 64: 861–866. 10.1016/0092-8674(91)90514-Y
- ↵Benjamini Y, Speed TP. 2012. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res 40: e72. 10.1093/nar/gks001
- ↵Bettencourt C, Hensman-Moss D, Flower M, Wiethoff S, Brice A, Goizet C, Stevanin G, Koutsis G, Karadima G, Panas M, 2016. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann Neurol 79: 983–990. 10.1002/ana.24656
- ↵Beyter D, Ingimundardottir H, Oddsson A, Eggertsson HP, Bjornsson E, Jonsson H, Atlason BA, Kristmundsdottir S, Mehringer S, Hardarson MT, 2021. Long-read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nat Genet 53: 779–786. 10.1038/s41588-021-00865-4
- Blauw HM, van Rheenen W, Koppers M, Van Damme P, Waibel S, Lemmens R, van Vught PWJ, Meyer T, Schulte C, Gasser T, 2012. NIPA1 polyalanine repeat expansions are associated with amyotrophic lateral sclerosis. Hum Mol Genet 21: 2497–2502. 10.1093/hmg/dds064
- ↵Bork P, Copley R. 2001. Filling in the gaps. Nature 409: 818–820. 10.1038/35057274
- ↵Botta A, Rossi G, Marcaurelio M, Fontana L, D'Apice MR, Brancati F, Massa R, Monckton DG, Sangiuolo F, Novelli G. 2017. Identification and characterization of 5′ CCG interruptions in complex DMPK expanded alleles. Eur J Hum Genet 25: 257–261. 10.1038/ejhg.2016.148
- Bragg DC, Mangkalaphiban K, Vaine CA, Kulkarni NJ, Shin D, Yadav R, Dhakal J, Ton M-L, Cheng A, Russo CT, 2017. Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1. Proc Natl Acad Sci 114: E11020–E11028. 10.1073/pnas.1712526114
- ↵Braida C, Stefanatos RKA, Adam B, Mahajan N, Smeets HJM, Niel F, Goizet C, Arveiler B, Koenig M, Lagier-Tourenne C, 2010. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum Mol Genet 19: 1399–1412. 10.1093/hmg/ddq015
- Brais B, Bouchard J-P, Xie Y-G, Rochefort DL, Chrétien N, Tomé FMS, Lafrentére RG, Rommens JM, Uyama E, Nohira O, 1998. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet 18: 164–167. 10.1038/ng0298-164
- ↵Breschel TS, McInnis MG, Margolis RL, Sirugo G, Corneliussen B, Simpson SG, McMahon FJ, MacKinnon DF, Xu JF, Pleasant N, 1997. A novel, heritable, expanding CTG repeat in an intron of the SEF2-1 gene on chromosome 18q21.1. Hum Mol Genet 6: 1855–1863. 10.1093/hmg/6.11.1855
- ↵Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion J-P, Hudson T, 1992. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68: 799–808. 10.1016/0092-8674(92)90154-5
- Brown SA, Warburton D, Brown LY, Yu C, Roeder ER, Stengel-Rutkowski S, Hennekam RCM, Muenke M. 1998. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet 20: 180–183. 10.1038/2484
- ↵Buijink AWG, Broersma M, van der Stouwe AMM, Sharifi S, Tijssen MAJ, Speelman JD, Maurits NM, van Rootselaar AF. 2016. Cerebellar atrophy in cortical myoclonic tremor and not in hereditary essential tremor—a voxel-based morphometry study. Cerebellum 15: 696–704. 10.1007/s12311-015-0734-0
- ↵Buxton J, Shelbourne P, Davies J, Jones C, Tongeren TV, Aslanidis C, de Jong P, Jansen G, Anvret M, Riley B, 1992. Detection of an unstable fragment of DNA specific to individuals with myotonic dystrophy. Nature 355: 547–548. 10.1038/355547a0
- Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, 1996. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271: 1423–1427. 10.1126/science.271.5254.1423
- ↵Cao Y, Tian R, Shi S, Du X, Jiao Y. 2021. Characterization and expression analysis of tandemly duplicated nicotinic acetylcholine receptors in pearl oysters after stimulation of pathogen-related molecular patterns. Comp Biochem Physiol B Biochem Mol Biol 256: 110615. 10.1016/j.cbpb.2021.110615
- ↵Caskey C, Pizzuti A, Fu Y, Fenwick R, Nelson D. 1992. Triplet repeat mutations in human disease. Science 256: 784–789. 10.1126/science.256.5058.784
- ↵Chamberlain NL, Driver ED, Miesfeld RL. 1994. The length and location of CAG trinucleotide repeats in the androgen receptor N-terminal domain affect transactivation function. Nucl Acids Res 22: 3181–3186. 10.1093/nar/22.15.3181
- ↵Chan EM, Young EJ, Ianzano L, Munteanu I, Zhao X, Christopoulos CC, Avanzini G, Elia M, Ackerley CA, Jovic NJ, 2003. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet 35: 125–127. 10.1038/ng1238
- Chandy KG, Fantino E, Wittekindt O, Kalman K, Tong L-L, Ho T-H, Gutman GA, Crocq M-A, Ganguli R, Nimgaonkar V, 1998. Isolation of a novel potassium channel gene hSKCa3 containing a polymorphic CAG repeat: a candidate for schizophrenia and bipolar disorder? Mol Psychiatry 3: 32–37. 10.1038/sj.mp.4000353
- ↵Chong SS, McCall AE, Cota J, Subramony SH, Orr HT, Hughes MR, Zoghbi HY. 1995. Gametic and somatic tissue–specific heterogeneity of the expanded SCA1 CAG repeat in spinocerebellar ataxia type 1. Nat Genet 10: 344–350. 10.1038/ng0795-344
- ↵Chung M, Ranum LPW, Duvick LA, Servadio A, Zoghbi HY, Orr HT. 1993. Evidence for a mechanism predisposing to intergenerational CAG repeat instability in spinocerebellar ataxia type I. Nat Genet 5: 254–258. 10.1038/ng1193-254
- ↵Ciosi M, Maxwell A, Cumming SA, Hensman Moss DJ, Alshammari AM, Flower MD, Durr A, Leavitt BR, Roos RAC, Holmans P, 2019. A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine 48: 568–580. 10.1016/j.ebiom.2019.09.020
- ↵Conforti FL, Spataro R, Sproviero W, Mazzei R, Cavalcanti F, Condino F, Simone IL, Logroscino G, Patitucci A, Magariello A, 2012. Ataxin-1 and ataxin-2 intermediate-length PolyQ expansions in amyotrophic lateral sclerosis. Neurology 79: 2315–2320. 10.1212/WNL.0b013e318278b618
- ↵Conway GS, Hettiarachchi S, Murray A, Jacobs PA. 1995. Fragile X premutations in familial premature ovarian failure. Lancet 346: 309–310. 10.1016/S0140-6736(95)92194-X
- ↵Corbett MA, Kroes T, Veneziano L, Bennett MF, Florian R, Schneider AL, Coppola A, Licchetta L, Franceschetti S, Suppa A, 2019. Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat Commun 10: 4920. 10.1038/s41467-019-12671-y
- ↵Corrado L, Mazzini L, Oggioni GD, Luciano B, Godi M, Brusco A, D'Alfonso S. 2011. ATXN-2 CAG repeat expansions are interrupted in ALS patients. Hum Genet 130: 575–580. 10.1007/s00439-011-1000-2
- ↵Correia F, Café C, Almeida J, Mouga S, Oliveira G. 2015. Autism spectrum disorder: FRAXE mutation, a rare etiology. J Autism Dev Disord 45: 888–892. 10.1007/s10803-014-2185-8
- ↵Cortese A, Simone R, Sullivan R, Vandrovcova J, Tariq H, Yau WY, Humphrey J, Jaunmuktane Z, Sivakumar P, Polke J, 2019. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet 51: 649–658. 10.1038/s41588-019-0372-4
- Course MM, Gudsnuk K, Smukowski SN, Winston K, Desai N, Ross JP, Sulovari A, Bourassa CV, Spiegelman D, Couthouis J, 2020. Evolution of a human-specific tandem repeat associated with ALS. Am J Hum Genet 107: 445–460. 10.1016/j.ajhg.2020.07.004
- ↵Craig-Holmes AP, Shaw MW. 1971. Polymorphism of human constitutive heterochromatin. Science 174: 702–704. 10.1126/science.174.4010.702
- ↵Cross-Disorder Group of the Psychiatric Genomics Consortium. 2013. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381: 1371–1379. 10.1016/S0140-6736(12)62129-1
- ↵Cumming SA, Hamilton MJ, Robb Y, Gregory H, McWilliam C, Cooper A, Adam B, McGhie J, Hamilton G, Herzyk P, 2018. De novo repeat interruptions are associated with reduced somatic instability and mild or absent clinical features in myotonic dystrophy type 1. Eur J Hum Genet 26: 1635–1647. 10.1038/s41431-018-0156-9
- ↵D'Andrea S, Pallotti F, Senofonte G, Castellini C, Paoli D, Lombardo F, Lenzi A, Francavilla S, Francavilla F, Barbonetti A. 2020. Polymorphic cytosine-adenine-guanine repeat length of androgen receptor gene and gender incongruence in trans women: a systematic review and meta-analysis of case-control studies. J Sex Med 17: 543–550. 10.1016/j.jsxm.2019.12.010
- ↵Das J, Lilleker J, Shereef H, Ealing J. 2017. Missense mutation in the ITPR1 gene presenting with ataxic cerebral palsy: description of an affected family and literature review. Neurol Neurochir Pol 51: 497–500. 10.1016/j.pjnns.2017.06.012
- ↵Dashnow H, Lek M, Phipson B, Halman A, Sadedin S, Lonsdale A, Davis M, Lamont P, Clayton JS, Laing NG, 2018. STRetch: detecting and discovering pathogenic short tandem repeat expansions. Genome Biol 19: 121. 10.1186/s13059-018-1505-2
- ↵De Antonio M, Dogan C, Hamroun D, Mati M, Zerrouki S, Eymard B, Katsahian S, Bassez G. 2016. Unravelling the myotonic dystrophy type 1 clinical spectrum: a systematic registry-based study with implications for disease classification. Rev Neurol (Paris) 172: 572–580. 10.1016/j.neurol.2016.08.003
- Debacker K, Winnepenninckx B, Longman C, Colgan J, Tolmie J, Murray R, van Luijk R, Scheers S, FitzPatrick D, Kooy F. 2007. The molecular basis of the folate-sensitive fragile site FRA11A at 11q13. Cytogenet Genome Res 119: 9–14. 10.1159/000109612
- De Baere E, Beysen D, Oley C, Lorenz B, Cocquet J, De Sutter P, Devriendt K, Dixon M, Fellous M, Fryns J-P, 2003. FOXL2 and BPES: mutational hotspots, phenotypic variability, and revision of the genotype-phenotype correlation. Am J Hum Genet 72: 478–487. 10.1086/346118
- ↵De Boulle K, Verkerk AJMH, Reyniers E, Vits L, Hendrickx J, Van Roy B, Van Den Bos F, de Graaff E, Oostra BA, Willems PJ. 1993. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat Genet 3: 31–35. 10.1038/ng0193-31
- Deere M, Sanford T, Francomano CA, Daniels K, Hecht JT. 1999. Identification of nine novel mutations in cartilage oligomeric matrix protein in patients with pseudoachondroplasia and multiple epiphyseal dysplasia. Am J Med Genet 85: 486–490. 10.1002/(SICI)1096-8628(19990827)85:5<486::AID-AJMG10>3.0.CO;2-O
- ↵DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, 2011. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72: 245–256. 10.1016/j.neuron.2011.09.011
- ↵DeJesus-Hernandez M, Aleff RA, Jackson JL, Finch NA, Baker MC, Gendron TF, Murray ME, McLaughlin IJ, Harting JR, Graff-Radford NR, 2021. Long-read targeted sequencing uncovers clinicopathological associations for C9orf72-linked diseases. Brain 144: 1082–1088. 10.1093/brain/awab006
- ↵Demaerel W, Mostovoy Y, Yilmaz F, Vervoort L, Pastor S, Hestand MS, Swillen A, Vergaelen E, Geiger EA, Coughlin CR, 2019. The 22q11 low copy repeats are characterized by unprecedented size and structural variability. Genome Res 29: 1389–1401. 10.1101/gr.248682.119
- Deng J, Yu J, Li P, Luan X, Cao L, Zhao J, Yu M, Zhang W, Lv H, Xie Z, 2020. Expansion of GGC repeat in GIPC1 is associated with oculopharyngodistal myopathy. Am J Hum Genet 106: 793–804. 10.1016/j.ajhg.2020.04.011
- De Roeck A, Duchateau L, Van Dongen J, Cacace R, Bjerke M, Van den Bossche T, Cras P, Vandenberghe R, De Deyn PP, Engelborghs S, 2018. An intronic VNTR affects splicing of ABCA7 and increases risk of Alzheimer's disease. Acta Neuropathol 135: 827–837. 10.1007/s00401-018-1841-z
- ↵De Roeck A, De Coster W, Bossaerts L, Cacace R, De Pooter T, Van Dongen J, D'Hert S, De Rijk P, Strazisar M, Van Broeckhoven C, 2019. NanoSatellite: accurate characterization of expanded tandem repeat length and sequence through whole genome long-read sequencing on PromethION. Genome Biol 20: 239. 10.1186/s13059-019-1856-3
- ↵Deschauer M, Hengel H, Rupprich K, Kreiß M, Schlotter-Weigel B, Grimmel M, Admard J, Schneider I, Alhaddad B, Gazou A, 2021. Bi-allelic truncating mutations in VWA1 cause neuromyopathy. Brain 144: 574–583. 10.1093/brain/awaa418
- ↵Deshmukh AD, Caron MC, Mohiuddin M, Lanni S, Panigrahi GB, Khan M, Engchuan W, Shum N, Faruqui A, Wang P, 2021. FAN1 exo- not endo-nuclease pausing on disease-associated slipped-DNA repeats: a mechanism of repeat instability. Cell Rep 37: 110078. 10.1016/j.celrep.2021.110078
- Devys D, Biancalana V, Rousseau F, Boué J, Mandel JL, Oberlé I. 1992. Analysis of full fragile X mutations in fetal tissues and monozygotic twins indicate that abnormal methylation and somatic heterogeneity are established early in development. Am J Med Genet 43: 208–216. 10.1002/ajmg.1320430134
- ↵Di Fabio F, Alvarado C, Gologan A, Youssef E, Voda L, Mitmaker E, Beitel LK, Gordon PH, Trifiro M. 2009. Somatic mosaicism of androgen receptor CAG repeats in colorectal carcinoma epithelial cells from men. J Surg Res 154: 38–44. 10.1016/j.jss.2008.05.013
- ↵Doi K, Monjo T, Hoang PH, Yoshimura J, Yurino H, Mitsui J, Ishiura H, Takahashi Y, Ichikawa Y, Goto J, 2014. Rapid detection of expanded short tandem repeats in personal genomics using hybrid sequencing. Bioinformatics 30: 815–822. 10.1093/bioinformatics/btt647
- ↵Dolzhenko E, van Vugt JJFA, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, Ajay SS, Rajan V, Lajoie BR, Johnson NH, 2017. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res 27: 1895–1903. 10.1101/gr.225672.117
- ↵Dolzhenko E, Deshpande V, Schlesinger F, Krusche P, Petrovski R, Chen S, Emig-Agius D, Gross A, Narzisi G, Bowman B, 2019. ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 35: 4754–4756. 10.1093/bioinformatics/btz431
- ↵Dolzhenko E, Bennett MF, Richmond PA, Trost B, Chen S, van Vugt JJFA, Nguyen C, Narzisi G, Gainullin VG, Gross AM, 2020. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol 21: 102. 10.1186/s13059-020-02017-z
- ↵Ebbert MTW, Farrugia SL, Sens JP, Jansen-West K, Gendron TF, Prudencio M, McLaughlin IJ, Bowman B, Seetin M, DeJesus-Hernandez M, 2018. Long-read sequencing across the C9orf72 ‘GGGGCC’ repeat expansion: implications for clinical use and genetic discovery efforts in human disease. Mol Neurodegener 13: 46. 10.1186/s13024-018-0274-4
- ↵Echenne B, Bassez G. 2013. Congenital and infantile myotonic dystrophy. In Handbook of Clinical Neurology (ed. Dulac O, ), Vol. 113, pp. 1387–1393. Elsevier, Amsterdam. https://linkinghub.elsevier.com/retrieve/pii/B9780444595652000095 (Accessed November 20, 2021). 10.1016/B978-0-444-59565-2.00009-5
- ↵Edwards A, Civitello A, Hammond HA, Caskey CT. 1991. DNA typing and genetic mapping with trimeric and tetrameric tandem repeats. Am J Hum Genet 49: 746–756.
- ↵Edwards A, Hammond HA, Jin L, Caskey CT, Chakraborty R. 1992. Genetic variation at five trimeric and tetrameric tandem repeat loci in four human population groups. Genomics 12: 241–253. 10.1016/0888-7543(92)90371-X
- ↵Eichler EE. 2001. Segmental duplications: what's missing, misassigned, and misassembled—and should we care?. Genome Res 11: 653–656. 10.1101/gr.188901
- ↵Eichler EE, Holden JJA, Popovich BW, Reiss AL, Snow K, Thibodeau SN, Richards CS, Ward PA, Nelson DL. 1994. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat Genet 8: 88–94. 10.1038/ng0994-88
- ↵Eisfeldt J, Mårtensson G, Ameur A, Nilsson D, Lindstrand A. 2020. Discovery of novel sequences in 1,000 Swedish genomes. Mol Biol Evol 37: 18–30. 10.1093/molbev/msz176
- ↵Elden AC, Kim H-J, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, 2010. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466: 1069–1075. 10.1038/nature09320
- ↵Faust I, Böker KO, Lichtenberg C, Kuhn J, Knabbe C, Hendig D. 2014. First description of the complete human xylosyltransferase-I promoter region. BMC Genet 15: 129. 10.1186/s12863-014-0129-0
- ↵Fautsch MP, Wieben ED, Baratz KH, Bhattacharyya N, Sadan AN, Hafford-Tear NJ, Tuft SJ, Davidson AE. 2021. TCF4-mediated Fuchs endothelial corneal dystrophy: insights into a common trinucleotide repeat-associated disease. Prog Retin Eye Res 81: 100883. 10.1016/j.preteyeres.2020.100883
- Favaro FP, Alvizi L, Zechi-Ceide RM, Bertola D, Felix TM, de Souza J, Raskin S, Twigg SRF, Weiner AMJ, Armas P, 2014. A noncoding expansion in EIF4A3 causes richieri-costa-pereira syndrome, a craniofacial disorder associated with limb defects. Am J Hum Genet 94: 120–128. 10.1016/j.ajhg.2013.11.020
- ↵Felbor U, Feichtinger W, Schmid M. 2003. The rare human fragile site 16B. Cytogenet Genome Res 100: 85–88. 10.1159/000072841
- ↵Ferro P, Catalano MG, Dell'Eva R, Fortunati N, Pfeffer U. 2002. The androgen receptor CAG repeat: a modifier of carcinogenesis? Mol Cell Endocrinol 193: 109–120. 10.1016/S0303-7207(02)00104-1
- ↵Florian RT, Kraft F, Leitão E, Kaya S, Klebe S, Magnin E, van Rootselaar A-F, Buratti J, Kühnel T, Schröder C, 2019. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with familial adult myoclonic epilepsy type 3. Nat Commun 10: 4919. 10.1038/s41467-019-12763-9
- ↵Flower M, Lomeikaite V, Ciosi M, Cumming S, Morales F, Lo K, Hensman Moss D, Jones L, Holmans P, Monckton DG, 2019. MSH3 modifies somatic instability and disease severity in Huntington's and myotonic dystrophy type 1. Brain 142: 1876–1886. 10.1093/brain/awz115
- ↵Fondon JW, Garner HR. 2004. Molecular origins of rapid and continuous morphological evolution. Proc Natl Acad Sci 101: 18058–18063. 10.1073/pnas.0408118101
- ↵Forman OP, De Risio L, Matiasek K, Platt S, Mellersh C. 2015. Spinocerebellar ataxia in the Italian Spinone dog is associated with an intronic GAA repeat expansion in ITPR1. Mamm Genome 26: 108–117. 10.1007/s00335-014-9547-6
- ↵Fotsing SF, Margoliash J, Wang C, Saini S, Yanicky R, Shleizer-Burko S, Goren A, Gymrek M. 2019. The impact of short tandem repeat variation on gene expression. Nat Genet 51: 1652–1659. 10.1038/s41588-019-0521-9
- ↵Fournier C, Anquetil V, Camuzat A, Stirati-Buron S, Sazdovitch V, Molina-Porcel L, Turbant S, Rinaldi D, Sánchez-Valle R, Barbier M, 2018. Interrupted CAG expansions in ATXN2 gene expand the genetic spectrum of frontotemporal dementias. Acta Neuropathol Commun 6: 41. 10.1186/s40478-018-0547-8
- ↵Franich NR, Hickey MA, Zhu C, Osborne GF, Ali N, Chu T, Bove NH, Lemesre V, Lerner RP, Zeitlin SO, 2019. Phenotype onset in Huntington's disease knock-in mice is correlated with the incomplete splicing of the mutant huntingtin gene. J Neurosci Res 97: 1590–1605. 10.1002/jnr.24493
- ↵Fredi M, Cavazzana I, Biasiotto G, Filosto M, Padovani A, Monti E, Tincani A, Franceschini F, Zanella I. 2019. C9orf72 intermediate alleles in patients with amyotrophic lateral sclerosis, systemic lupus erythematosus, and rheumatoid arthritis. Neuromolecular Med 21: 150–159. 10.1007/s12017-019-08528-8
- ↵Fu Y-H, Kuhl DPA, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkert AJMH, Holden JJA, Fenwick RG, Warren ST, 1991. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 67: 1047–1058. 10.1016/0092-8674(91)90283-5
- ↵Fu YH, Pizzuti A, Fenwick RG, King J, Rajnarayan S, Dunne PW, Dubel J, Nasser GA, Ashizawa T, de Jong P, 1992. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 255: 1256–1258. 10.1126/science.1546326
- ↵Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N, 2011. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med 17: 720–725. 10.1038/nm.2374
- ↵Garg P, Jadhav B, Rodriguez OL, Patel N, Martin-Trujillo A, Jain M, Metsu S, Olsen H, Paten B, Ritz B, 2020. A survey of rare epigenetic variation in 23,116 human genomes identifies disease-relevant epivariations and CGG expansions. Am J Hum Genet 107: 654–669. 10.1016/j.ajhg.2020.08.019
- ↵Garza R, Gill AJ, Bastien BL, Garcia-Mesa Y, Gruenewald AL, Gelman BB, Tsima B, Gross R, Letendre SL, Kolson DL. 2020. Heme oxygenase-1 promoter (GT)n polymorphism associates with HIV neurocognitive impairment. Neurol Neuroimmunol Neuroinflamm 7: e710. 10.1212/NXI.0000000000000710
- ↵Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium, Lee J-M, Correia K, Loupe J, Kim K-H, Barker D, Hong EP, Chao MJ, Long JD, Lucente D, 2019. CAG repeat not polyglutamine length determines timing of Huntington's disease onset. Cell 178: 887–900.e14. 10.1016/j.cell.2019.06.036
- ↵Gentalen E, Chee M. 1999. A novel method for determining linkage between DNA sequences: hybridization to paired probe arrays. Nucleic Acids Res 27: 1485–1491. 10.1093/nar/27.6.1485
- ↵Giesselmann P, Brändl B, Raimondeau E, Bowen R, Rohrandt C, Tandon R, Kretzmer H, Assum G, Galonska C, Siebert R, 2019. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat Biotechnol 37: 1478–1481. 10.1038/s41587-019-0293-x
- ↵Gill AJ, Garza R, Ambegaokar SS, Gelman BB, Kolson DL. 2018. Heme oxygenase-1 promoter region (GT)n polymorphism associates with increased neuroimmune activation and risk for encephalitis in HIV infection. J Neuroinflammation 15: 70. 10.1186/s12974-018-1102-z
- ↵Glineburg MR, Todd PK, Charlet-Berguerand N, Sellier C. 2018. Repeat-associated non-AUG (RAN) translation and other molecular mechanisms in fragile X tremor ataxia syndrome. Brain Res 1693: 43–54. 10.1016/j.brainres.2018.02.006
- Goodman FR, Bacchelli C, Brady AF, Brueton LA, Fryns J-P, Mortlock DP, Innis JW, Holmes LB, Donnenfeld AE, Feingold M, 2000. Novel HOXA13 mutations and the phenotypic spectrum of hand-foot-genital syndrome. Am J Hum Genet 67: 197–202. 10.1086/302961
- ↵Gosden JR, Mitchell AR, Buckland RA, Clayton RP, Evans HJ. 1975. The location of four human satellite DNAs on human chromosomes. Cytogenet Genome Res 14: 338–339. 10.1159/000130376
- ↵Gosden JR, Spowart G, Lawrie SS. 1981. Satellite DNA and cytological staining patterns in heterochromatic inversions of human chromosome 9. Hum Genet 58: 276–278. 10.1007/BF00294922
- ↵Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, Pallesen J, Agerbo E, Andreassen OA, Anney R, 2019. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 51: 431–444. 10.1038/s41588-019-0344-8
- ↵Grunseich C, Kats IR, Bott LC, Rinaldi C, Kokkinis A, Fox D, Chen K, Schindler AB, Mankodi AK, Shrader JA, 2014. Early onset and novel features in a spinal and bulbar muscular atrophy patient with a 68 CAG repeat. Neuromuscul Disord 24: 978–981. 10.1016/j.nmd.2014.06.441
- Gu Y, Shen Y, Gibbs RA, Nelson DL. 1996. Identification of FMR2, a novel gene associated with the FRAXE CCG repeat and CpG island. Nat Genet 13: 109–113. 10.1038/ng0596-109
- ↵Guo M, Li S, Zhou Y, Li M, Wen Z. 2021. Comparative analysis for the performance of long-read-based structural variation detection pipelines in tandem repeat regions. Front Pharmacol 12: 658072. 10.3389/fphar.2021.658072
- ↵Gymrek M, Golan D, Rosset S, Erlich Y. 2012. lobSTR: a short tandem repeat profiler for personal genomes. Genome Res 22: 1154–1162. 10.1101/gr.135780.111
- ↵Gymrek M, Willems T, Guilmatre A, Zeng H, Markus B, Georgiev S, Daly MJ, Price AL, Pritchard JK, Sharp AJ, 2016. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat Genet 48: 22–29. 10.1038/ng.3461
- ↵Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, Grigsby J, Gage B, Hagerman PJ. 2001. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 57: 127–130. 10.1212/WNL.57.1.127
- ↵Han Z, Hu Y, Lv Y, Rose JKC, Sun Y, Shen F, Wang Y, Zhang X, Xu X, Wu T, 2018. Natural variation underlies differences in ETHYLENE RESPONSE FACTOR17 activity in fruit peel degreening. Plant Physiol 176: 2292–2304. 10.1104/pp.17.01320
- ↵Handt O, Sutherland GR, Richards RI. 2000. Fragile sites and minisatellite repeat instability. Mol Genet Metab 70: 99–105. 10.1006/mgme.2000.2996
- ↵Hannan AJ. 2010. Tandem repeat polymorphisms: modulators of disease susceptibility and candidates for ‘missing heritability’. Trends Genet 26: 59–65. 10.1016/j.tig.2009.11.008
- ↵Hannan AJ. 2018. Tandem repeats mediating genetic plasticity in health and disease. Nat Rev Genet 19: 286–298. 10.1038/nrg.2017.115
- ↵Hare L, Bernard P, Sánchez FJ, Baird PN, Vilain E, Kennedy T, Harley VR. 2009. Androgen receptor repeat length polymorphism associated with male-to-female transsexualism. Biol Psychiatry 65: 93–96. 10.1016/j.biopsych.2008.08.033
- ↵Harley HG, Brook JD, Rundle SA, Crow S, Reardon W, Buckler AJ, Harper PS, Housman DE, Shaw DJ. 1992. Expansion of an unstable DNA region and phenotypic variation in myotonic dystrophy. Nature 355: 545–546. 10.1038/355545a0
- ↵Harper PS, Harley HG, Reardon W, Shaw DJ. 1992. Anticipation in myotonic dystrophy: new light on an old problem. Am J Hum Genet 51: 10–16.
- ↵Hauser J, Kapelski P, Czerski PM, Godlewski S, Dmitrzak-Weglarz M, Twardowska K, Rybakowski JK. 2002. [Lack of association between VNTR polymorphism of DAT gene and schizophrenia]. Psychiatr Pol 36: 403–412.
- ↵Heitz D, Rousseau F, Devys D, Saccone S, Abderrahim H, Le Paslier D, Cohen D, Vincent A, Toniolo D, Della Valle G, 1991. Isolation of sequences that span the fragile X and identification of a fragile X-related CpG island. Science 251: 1236–1239. 10.1126/science.2006411
- ↵Hewett DR, Handt O, Hobson L, Mangelsdorf M, Eyre HJ, Baker E, Sutherland GR, Schuffenhauer S, Mao J, Richards RI. 1998. FRA10B structure reveals common elements in repeat expansion and chromosomal fragile site genesis. Mol Cell 1: 773–781. 10.1016/S1097-2765(00)80077-5
- ↵Hirst M, Grewal P, Flannery A, Slatter R, Maher E, Barton D, Fryns JP, Davies K. 1995. Two new cases of FMR1 deletion associated with mental impairment. Am J Hum Genet 56: 67–74. 10.1002/ajmg.1320560115
- ↵Hocking T, Feichtinger W, Schmid M, Haan EA, Baker E, Sutherland GR. 1999. Homozygotes for FRA16B are normal. Chromosome Res 7: 553–556. 10.1023/A:1009293613064
- ↵Holmes SE, O'Hearn EE, McInnis MG, Gorelick-Feldman DA, Kleiderlein JJ, Callahan C, Kwak NG, Ingersoll-Ashworth RG, Sherr M, Sumner AJ, 1999. Expansion of a novel CAG trinucleotide repeat in the 5′ region of PPP2R2B is associated with SCA12. Nat Genet 23: 391–392. 10.1038/70493
- ↵Holmes SE, O'Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, Fleisher A, Stevanin G, Brice A, Potter NT, 2001. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease–like 2. Nat Genet 29: 377–378. 10.1038/ng760
- ↵Hommelsheim CM, Frantzeskakis L, Huang M, Ülker B. 2015. PCR amplification of repetitive DNA: a limitation to genome editing technologies and many other applications. Sci Rep 4: 5052. 10.1038/srep05052
- ↵Hori T, Takahashi E, Tsuji H, Tsuji S, Murata M. 1988. Fragile X expression in thymidine-prototrophic and auxotrophic human-mouse somatic cell hybrids under low and high thymidylate stress conditions. Cytogenet Cell Genet 47: 177–180. 10.1159/000132543
- ↵Howard-Peebles PN. 1980. Fragile sites in human chromosomes II: demonstration of the fragile site Xq27 in carriers of X-linked mental retardation. Am J Med Genet 7: 497–501. 10.1002/ajmg.1320070410
- ↵Höweler CJ, Busch HFM, Geraedts JPM, Niermeijer MF, Staal A. 1989. Anticipation in myotonic dystrophy: fact or fiction? Brain 112: 779–797. 10.1093/brain/112.3.779
- ↵Hoyt SJ, Storer JM, Hartley GA, Grady PGS, Gershman A, de Lima LG, Limouse C, Halabian R, Wojenski L, Rodriguez M, 2021. From telomere to telomere: the transcriptional and epigenetic state of human repeat elements. bioRxiv 10.1101/2021.07.12.451456 (Accessed November 20, 2021)
- ↵The Huntington's Disease Collaborative Research Group. 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971–983. 10.1016/0092-8674(93)90585-E
- Imbert G, Trottier Y, Beckmann J, Mandel JL. 1994. The gene for the TATA binding protein (TBP) that contains a highly polymorphic protein coding CAG repeat maps to 6q27. Genomics 21: 667–668. 10.1006/geno.1994.1335
- Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, Garnier J-M, Weber C, Mandel J-L, Cancel G, Abbas N, 1996. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet 14: 285–291. 10.1038/ng1196-285
- ↵Irony-Tur Sinai M, Kerem B. 2019. Genomic instability in fragile sites—still adding the pieces. Genes Chromosomes Cancer 58: 295–304. 10.1002/gcc.22715
- ↵Ishiguro T, Sato N, Ueyama M, Fujikake N, Sellier C, Kanegami A, Tokuda E, Zamiri B, Gall-Duncan T, Mirceta M, 2017. Regulatory role of RNA chaperone TDP-43 for RNA misfolding and repeat-associated translation in SCA31. Neuron 94: 108–124.e7. 10.1016/j.neuron.2017.02.046
- ↵Ishiura H, Tsuji S. 2020. Advances in repeat expansion diseases and a new concept of repeat motif–phenotype correlation. Curr Opin Genet Dev 65: 176–185. 10.1016/j.gde.2020.05.029
- ↵Ishiura H, Doi K, Mitsui J, Yoshimura J, Matsukawa MK, Fujiyama A, Toyoshima Y, Kakita A, Takahashi H, Suzuki Y, 2018. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat Genet 50: 581–590. 10.1038/s41588-018-0067-2
- ↵Ishiura H, Shibata S, Yoshimura J, Suzuki Y, Qu W, Doi K, Almansour MA, Kikuchi JK, Taira M, Mitsui J, 2019. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat Genet 51: 1222–1232. 10.1038/s41588-019-0458-z
- ↵Ivashchenko DV, Shuvalov SA, Chuprova NA, Kibitov AO. 2015. The association of polymorphisms in DAT (40 bp VNTR, C>T 3′UTR) and DBH (−1021 C/T) genes with the severe complications of alcohol withdrawal state. Psychiatr Genet 25: 268–269. 10.1097/YPG.0000000000000101
- ↵Jacquemont S, Hagerman R, Leehey M, Hall DA, Levine RA, Brunberg JA, Zhang L, Jardini T, Gane LW, Harris SW, 2004. Penetrance of the fragile X–associated tremor/ataxia syndrome in a premutation carrier population. JAMA 291: 460–469. 10.1001/jama.291.4.460
- ↵Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, 2018a. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol 36: 338–345. 10.1038/nbt.4060
- ↵Jain M, Olsen HE, Turner DJ, Stoddart D, Bulazel KV, Paten B, Haussler D, Willard HF, Akeson M, Miga KH. 2018b. Linear assembly of a human centromere on the Y chromosome. Nat Biotechnol 36: 321–323. 10.1038/nbt.4109
- ↵Jiao W-B, Accinelli GG, Hartwig B, Kiefer C, Baker D, Severing E, Willing E-M, Piednoel M, Woetzel S, Madrid-Herrero E, 2017. Improving and correcting the contiguity of long-read genome assemblies of three plant species using optical mapping and chromosome conformation capture data. Genome Res 27: 778–786. 10.1101/gr.213652.116
- Jones C, Slljepcevic P, Marsh S, Baker E, Langdon WY, Richards RI, Tunnacliffe A. 1994. Physical linkage of the fragile site FRA11B and a Jacobsen Syndrome chromosome deletion breakpoint in 11q23. 3. Hum Mol Genet 3: 2123–2130. 10.1093/hmg/3.12.2123
- ↵Joosten IBT, Hellebrekers DMEI, de Greef BTA, Smeets HJM, de Die-Smulders CEM, Faber CG, Gerrits MM. 2020. Parental repeat length instability in myotonic dystrophy type 1 pre- and protomutations. Eur J Hum Genet 28: 956–962. 10.1038/s41431-020-0601-4
- ↵Katsumata Y, Fardo DW, Bachstetter AD, Artiushin SC, Wang W-X, Wei A, Brzezinski LJ, Nelson BG, Huang Q, Abner EL, 2020. Alzheimer disease pathology-associated polymorphism in a complex variable number of tandem repeat region within the MUC6 gene, near the AP2A2 gene. J Neuropathol Exp Neurol 79: 3–21. 10.1093/jnen/nlz116
- ↵Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M, Akiguchi I, 1994. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 8: 221–228. 10.1038/ng1194-221
- ↵Kazemi-Esfarjani P, Trifiro MA, Pinsky L. 1995. Evidence for a repressive function of the long polyglutamine tract in the human androgen receptor: possible pathogenetic relevance for the (CAG)n-expanded neuronopathies. Hum Mol Genet 4: 523–527. 10.1093/hmg/4.4.523
- ↵Kehl A, Cizinauskas S, Langbein-Detsch I, Mueller E. 2019. NHLRC1 dodecamer expansion in a Welsh Corgi (Pembroke) with Lafora disease. Anim Genet 50: 413–414. 10.1111/age.12795
- ↵Kiełbasa SM, Wan R, Sato K, Horton P, Frith MC. 2011. Adaptive seeds tame genomic sequence comparison. Genome Res 21: 487–493. 10.1101/gr.113985.110
- ↵Knight SJL, Flannery AV, Hirst MC, Campbell L, Christodoulou Z, Phelps SR, Pointon J, Middleton-Price HR, Barnicoat A, Pembrey ME, 1993. Trinucleotide repeat amplification and hypermethylation of a CpG island in FRAXE mental retardation. Cell 74: 127–134. 10.1016/0092-8674(93)90300-F
- ↵Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, Habu T, Liu W, Okuda H, Koizumi A. 2011. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet 89: 121–130. 10.1016/j.ajhg.2011.05.015
- ↵Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K, Takahashi H, Kondo R, Ishikawa A, Hayashi T, 1994. Unstable expansion of CAG repeat in hereditary dentatorubral–pallidoluysian atrophy (DRPLA). Nat Genet 6: 9–13. 10.1038/ng0194-9
- Koide R, Kobayashi S, Shimohata T, Ikeuchi T, Maruyama M, Saito M, Yamada M, Takahashi H, Tsuji S. 1999. A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum Mol Genet 8: 2047–2053. 10.1093/hmg/8.11.2047
- ↵Koob MD, Benzow KA, Bird TD, Day JW, Moseley ML, Ranum LPW. 1998. Rapid cloning of expanded trinucleotide repeat sequences from genomic DNA. Nat Genet 18: 72–75. 10.1038/ng0198-72
- ↵Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, Ranum LPW. 1999. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat Genet 21: 379–384. 10.1038/7710
- ↵Kozarewa I, Turner DJ. 2011. Amplification-free library preparation for paired-end illumina sequencing. Methods Mol Biol 733: 257–266. 10.1007/978-1-61779-089-8_18
- ↵Kozarewa I, Ning Z, Quail MA, Sanders MJ, Berriman M, Turner DJ. 2009. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G + C)-biased genomes. Nat Methods 6: 291–295. 10.1038/nmeth.1311
- ↵Krans A, Skariah G, Zhang Y, Bayly B, Todd PK. 2019. Neuropathology of RAN translation proteins in fragile X-associated tremor/ataxia syndrome. Acta Neuropathol Commun 7: 152. 10.1186/s40478-019-0782-7
- ↵Kremer EJ, Pritchard M, Lynch M, Yu S, Holman K, Baker E, Warren ST, Schlessinger D, Sutherland GR, Richards RI. 1991. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence P(CCG)n. Science 252: 1711–1714. 10.1126/science.1675488
- ↵Laabs B-H, Klein C, Pozojevic J, Domingo A, Brüggemann N, Grütz K, Rosales RL, Jamora RD, Saranza G, Diesta CCE, 2021. Identifying genetic modifiers of age-associated penetrance in X-linked dystonia-parkinsonism. Nat Commun 12: 3216. 10.1038/s41467-021-23491-4
- ↵LaCroix AJ, Stabley D, Sahraoui R, Adam MP, Mehaffey M, Kernan K, Myers CT, Fagerstrom C, Anadiotis G, Akkari YM, 2019. GGC repeat expansion and exon 1 methylation of XYLT1 is a common pathogenic variant in Baratela-Scott syndrome. Am J Hum Genet 104: 35–44. 10.1016/j.ajhg.2018.11.005
- ↵Lafreniére RG, Rochefort DL, Chrétien N, Rommens JM, Cochius JI, Kälviäinen R, Nousiainen U, Patry G, Farrell K, Söderfeldt B, 1997. Unstable insertion in the 5′ flanking region of the cystatin B gene is the most common mutation in progressive myoclonus epilepsy type 1, EPM1. Nat Genet 15: 298–302. 10.1038/ng0397-298
- ↵Lafuente A, Bernardo M, Mas S, Crescenti A, Aparici M, Gasso P, Catalan R, Mateos J, Lomena F, Parellada E. 2007. Dopamine transporter (DAT) genotype (VNTR) and phenotype in extrapyramidal symptoms induced by antipsychotics. Schizophr Res 90: 115–122. 10.1016/j.schres.2006.09.031
- ↵Lalioti MD, Scott HS, Buresi C, Rossier C, Bottani A, Morris MA, Malafosse A, Antonarakis SE. 1997. Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature 386: 847–851. 10.1038/386847a0
- ↵La Spada ARL, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. 1991. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352: 77–79. 10.1038/352077a0
- ↵Latham GJ, Coppinger J, Hadd AG, Nolin SL. 2014. The role of AGG interruptions in fragile X repeat expansions: a twenty-year perspective. Front Genet 5: 244. 10.3389/fgene.2014.00244
- ↵Ledbetter DH, Ledbetter SA, Nussbaum RL. 1986. Implications of fragile X expression in normal males for the nature of the mutation. Nature 324: 161–163. 10.1038/324161a0
- ↵Li Q, Li Y, Song J, Xu H, Xu J, Zhu Y, Li X, Gao H, Dong L, Qian J, 2014. High-accuracy de novo assembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy. New Phytol 204: 1041–1049. 10.1111/nph.12966
- ↵Li H, Dawood M, Khayat MM, Farek JR, Jhangiani SN, Khan ZM, Mitani T, Coban-Akdemir Z, Lupski JR, Venner E, 2021. Exome variant discrepancies due to reference-genome differences. Am J Hum Genet 108: 1239–1250. 10.1016/j.ajhg.2021.05.011
- ↵Lin C-H, Chen C-M, Hou Y-T, Wu Y-R, Hsieh-Li H-M, Su M-T, Lee-Chen G-J. 2010. The CAG repeat in SCA12 functions as a cis element to up-regulate PPP2R2B expression. Hum Genet 128: 205–212. 10.1007/s00439-010-0843-2
- ↵Lindblad K, Savontaus ML, Stevanin G, Holmberg M, Digre K, Zander C, Ehrsson H, David G, Benomar A, Nikoskelainen E, 1996. An expanded CAG repeat sequence in spinocerebellar ataxia type 7. Genome Res 6: 965–971. 10.1101/gr.6.10.965
- ↵Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LPW. 2001. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293: 864–867. 10.1126/science.1062125
- ↵Lit L, Belanger JM, Boehm D, Lybarger N, Oberbauer AM. 2013. Differences in behavior and activity associated with a Poly(A) expansion in the dopamine transporter in belgian malinois. PLoS One 8: e82948. 10.1371/journal.pone.0082948
- ↵Liu Q, Zhang P, Wang D, Gu W, Wang K. 2017. Interrogating the “unsequenceable” genomic trinucleotide repeat disorders by long-read sequencing. Genome Med 9: 65. 10.1186/s13073-017-0456-7
- ↵Loesch DZ, Hay DA, Mulley J. 1994. Transmitting males and carrier females in fragile X-revisited. Am J Med Genet 51: 392–399. 10.1002/ajmg.1320510418
- ↵Logsdon GA, Vollger MR, Hsieh P, Mao Y, Liskovykh MA, Koren S, Nurk S, Mercuri L, Dishuck PC, Rhie A, 2021. The structure, function and evolution of a complete human chromosome 8. Nature 593: 101–107. 10.1038/s41586-021-03420-7
- ↵Lohi H, Young EJ, Fitzmaurice SN, Rusbridge C, Chan EM, Vervoort M, Turnbull J, Zhao X-C, Ianzano L, Paterson AD, 2005. Expanded repeat in canine epilepsy. Science 307: 81. 10.1126/science.1102832
- ↵López Castel A, Cleary JD, Pearson CE. 2010. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol 11: 165–170. 10.1038/nrm2854
- ↵Lu T-Y, The Human Genome Structural Variation Consortium, Chaisson MJP. 2021. Profiling variable-number tandem repeat variation across populations using repeat-pangenome graphs. Nat Commun 12: 4250. 10.1038/s41467-021-24378-0
- ↵Lubahn DB, Joseph DR, Sar M, Tan J, Higgs HN, Larson RE, French FS, Wilson EM. 1988. The human androgen receptor: complementary deoxyribonucleic acid cloning, sequence analysis and gene expression in prostate. Mol Endocrinol 2: 1265–1275. 10.1210/mend-2-12-1265
- ↵Lubs HA. 1969. A marker X chromosome. Am J Hum Genet 21: 231–244.
- ↵Luciani JJ, Depetris D, Missirian C, Mignon-Ravix C, Metzler-Guillemain C, Megarbane A, Moncla A, Mattei M-G. 2005. Subcellular distribution of HP1 proteins is altered in ICF syndrome. Eur J Hum Genet 13: 41–51. 10.1038/sj.ejhg.5201293
- ↵Lugenbeel KA, Peier AM, Carson NL, Chudley AE, Nelson DL. 1995. Intragenic loss of function mutations demonstrate the primary role of FMR1 in fragile X syndrome. Nat Genet 10: 483–485. 10.1038/ng0895-483
- ↵Madeira JLO, Souza ABC, Cunha FS, Batista RL, Gomes NL, Rodrigues AS, Mennucci de Haidar Jorge F, Chadi G, Callegaro D, Mendonca BB, 2018. A severe phenotype of Kennedy disease associated with a very large CAG repeat expansion. Muscle Nerve 57: E95–E97. 10.1002/mus.25952
- ↵Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barceló J, O'Hoy K, 1992. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science 255: 1253–1255. 10.1126/science.1546325
- ↵Mandel J-L. 1993. Questions of expansion. Nat Genet 4: 8–9. 10.1038/ng0593-8
- ↵Mankodi A, Urbinati CR, Yuan Q, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS, Thornton CA. 2001. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet 10: 2165–2170. 10.1093/hmg/10.19.2165
- ↵Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, Cannon SC, Thornton CA. 2002. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel Pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell 10: 35–44. 10.1016/S1097-2765(02)00563-4
- ↵Margolis RL, O'Hearn E, Rosenblatt A, Willour V, Holmes SE, Franz ML, Callahan C, Hwang HS, Troncoso JC, Ross CA. 2001. A disorder similar to Huntington's disease is associated with a novel CAG repeat expansion. Ann Neurol 50: 373–380. 10.1002/ana.1312
- ↵Martin JP, Bell J. 1943. A pedigree of mental defect showing sex-linkage. J Neurol Psychiatry 6: 154–157. 10.1136/jnnp.6.3-4.154
- ↵Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, Khajavi M, McCall AE, Davis CF, Zu L, 2000. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet 26: 191–194. 10.1038/79911
- ↵Metsu S, Rainger JK, Debacker K, Bernhard B, Rooms L, Grafodatskaya D, Weksberg R, Fombonne E, Taylor MS, Scherer SW, 2014a. A CGG-repeat expansion mutation in ZNF713 causes FRA7A: association with autistic spectrum disorder in two families. Hum Mutat 35: 1295–1300. 10.1002/humu.22683
- ↵Metsu S, Rooms L, Rainger J, Taylor MS, Bengani H, Wilson DI, Chilamakuri CSR, Morrison H, Vandeweyer G, Reyniers E, 2014b. FRA2A is a CGG repeat expansion associated with silencing of AFF3. PLoS Genet 10: e1004242. 10.1371/journal.pgen.1004242
- ↵Mhatre AN, Trifiro MA, Kaufman M, Kazemi-Esfarjani P, Figlewicz D, Rouleau G, Pinsky L. 1993. Reduced transcriptional regulatory competence of the androgen receptor in X–linked spinal and bulbar muscular atrophy. Nat Genet 5: 184–188. 10.1038/ng1093-184
- ↵Midha MK, Wu M, Chiu K-P. 2019. Long-read sequencing in deciphering human genetics to a greater depth. Hum Genet 138: 1201–1215. 10.1007/s00439-019-02064-y
- ↵Miga KH. 2015. Completing the human genome: the progress and challenge of satellite DNA assembly. Chromosome Res 23: 421–426. 10.1007/s10577-015-9488-2
- ↵Miga KH, Koren S, Rhie A, Vollger MR, Gershman A, Bzikadze A, Brooks S, Howe E, Porubsky D, Logsdon GA, 2020. Telomere-to-telomere assembly of a complete human X chromosome. Nature 585: 79–84. 10.1038/s41586-020-2547-7
- ↵Miller ZA, Sturm VE, Camsari GB, Karydas A, Yokoyama JS, Grinberg LT, Boxer AL, Rosen HJ, Rankin KP, Gorno-Tempini ML, 2016. Increased prevalence of autoimmune disease within C9 and FTD/MND cohorts: completing the picture. Neurol Neuroimmunol Neuroinflamm 3: e301. 10.1212/NXI.0000000000000301
- ↵Miller DE, Sulovari A, Wang T, Loucks H, Hoekzema K, Munson KM, Lewis AP, Fuerte EPA, Paschal CR, Walsh T, 2021. Targeted long-read sequencing identifies missing disease-causing variation. Am J Hum Genet 108: 1436–1449. 10.1016/j.ajhg.2021.06.006
- ↵Mitra I, Huang B, Mousavi N, Ma N, Lamkin M, Yanicky R, Shleizer-Burko S, Lohmueller KE, Gymrek M. 2021. Patterns of de novo tandem repeat mutations and their role in autism. Nature 589: 246–250. 10.1038/s41586-020-03078-7
- ↵Mitsuhashi S, Nakagawa S, Takahashi Ueda M, Imanishi T, Frith MC, Mitsuhashi H. 2017. Nanopore-based single molecule sequencing of the D4Z4 array responsible for facioscapulohumeral muscular dystrophy. Sci Rep 7: 14789. 10.1038/s41598-017-13712-6
- ↵Mitsuhashi S, Frith MC, Mizuguchi T, Miyatake S, Toyota T, Adachi H, Oma Y, Kino Y, Mitsuhashi H, Matsumoto N. 2019. Tandem-genotypes: robust detection of tandem repeat expansions from long DNA reads. Genome Biol 20: 58. 10.1186/s13059-019-1667-6
- ↵Mohsen-Pour N, Talebi T, Naderi N, Moghadam MH, Maleki M, Kalayinia S. 2021. Chromosome 9 inversion: pathogenic or benign? A comprehensive systematic review of all clinical reports. Curr Mol Med 21: 1–16. 10.2174/1566524021666210806161128
- ↵Mojarad BA, Yin Y, Manshaei R, Backstrom I, Costain G, Heung T, Merico D, Marshall CR, Bassett AS, Yuen RKC. 2021a. Genome sequencing broadens the range of contributing variants with clinical implications in schizophrenia. Transl Psychiatry 11: 84. 10.1038/s41398-021-01211-2
- ↵Mojarad BA, Engchuan W, Trost B, Backstrom I, Yin Y, Thiruvahindrapuram B, Pallotto L, Khan M, Pellecchia G, Haque B, 2021b. Genome-wide tandem repeat expansions contribute to schizophrenia risk. medRxiv 10.1101/2021.12.17.21267642
- ↵Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, Chen G, Weatherspoon MR, Clark HB, Ebner TJ, 2006. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet 38: 758–769. 10.1038/ng1827
- ↵Moss DJH, Pardiñas AF, Langbehn D, Lo K, Leavitt BR, Roos R, Durr A, Mead S, Holmans P, Jones L, 2017. Identification of genetic variants associated with Huntington's disease progression: a genome-wide association study. Lancet Neurol 16: 701–711. 10.1016/S1474-4422(17)30161-8
- ↵Mostacciuolo M, Pastorello E, Vazza G, Miorin M, Angelini C, Tomelleri G, Galluzzi G, Trevisan C. 2009. Facioscapulohumeral muscular dystrophy: epidemiological and molecular study in a north-east Italian population sample. Clin Genet 75: 550–555. 10.1111/j.1399-0004.2009.01158.x
- ↵Mousavi N, Shleizer-Burko S, Yanicky R, Gymrek M. 2019. Profiling the genome-wide landscape of tandem repeat expansions. Nucleic Acids Res 47: e90–e90. 10.1093/nar/gkz501
- ↵Mukamel RE, Handsaker RE, Sherman MA, Barton AR, Zheng Y, McCarroll SA, Loh P-R. 2021. Protein-coding repeat polymorphisms strongly shape diverse human phenotypes. Science 373: 1499–1505. 10.1126/science.abg8289
- Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, Lindhout D, Cole WG, Henn W, Knoll JHM, 1997. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 89: 773–779. 10.1016/S0092-8674(00)80260-3
- ↵Murray A, Webb J, Grimley S, Conway G, Jacobs P. 1998. Studies of FRAXA and FRAXE in women with premature ovarian failure. J Med Genet 35: 637–640. 10.1136/jmg.35.8.637
- ↵Musova Z, Mazanec R, Krepelova A, Ehler E, Vales J, Jaklova R, Prochazka T, Koukal P, Marikova T, Kraus J, 2009. Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am J Med Genet 149A: 1365–1374. 10.1002/ajmg.a.32987
- ↵Nagafuchi S, Yanagisawa H, Sato K, Shirayama T, Ohsaki E, Bundo M, Takeda T, Tadokoro K, Kondo I, Murayama N, 1994. Dentatorubral and pallidoluysian atrophy expansion of an unstable CAG trinucleotide on chromosome 12p. Nat Genet 6: 14–18. 10.1038/ng0194-14
- ↵Nancarrow JK, Kremer E, Holman K, Eyre H, Doggett NA, Le Paslier D, Callen DF, Sutherland GR, Richards RI. 1994. Implications of FRA16A structure for the mechanism of chromosomal fragile site genesis. Science 264: 1938–1941. 10.1126/science.8009225
- ↵Nelson PT, Fardo DW, Katsumata Y. 2020. The MUC6/AP2A2 locus and its relevance to Alzheimer's disease: a review. J Neuropathol Exp Neurol 79: 568–584. 10.1093/jnen/nlaa024
- ↵Neueder A, Landles C, Ghosh R, Howland D, Myers RH, Faull RLM, Tabrizi SJ, Bates GP. 2017. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington's disease patients. Sci Rep 7: 1307. 10.1038/s41598-017-01510-z
- ↵Neueder A, Dumas AA, Benjamin AC, Bates GP. 2018. Regulatory mechanisms of incomplete huntingtin mRNA splicing. Nat Commun 9: 3955. 10.1038/s41467-018-06281-3
- ↵Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, 2021. The complete sequence of a human genome. bioRxiv 10.1101/2021.05.26.445798 (Accessed November 20, 2021)
- ↵Nussbaum RL, Airhart SD, Ledbetter DH. 1986. Recombination and amplification of pyrimidine-rich sequences may be responsible for initiation and progression of the Xq27 fragile site: an hypothesis. Am J Med Genet 23: 715–721. 10.1002/ajmg.1320230162
- ↵Oberlé I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boué J, Bertheas MF, Mandel JL. 1991. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252: 1097–1102. 10.1126/science.252.5009.1097
- ↵Okubo M, Doi H, Fukai R, Fujita A, Mitsuhashi S, Hashiguchi S, Kishida H, Ueda N, Morihara K, Ogasawara A, 2019. GGC repeat expansion of NOTCH2NLC in adult patients with leukoencephalopathy. Ann Neurol 86: 962–968. 10.1002/ana.25586
- ↵Oostra BA, Verkerk AJMH. 1992. Review: the fragile X syndrome: isolation of the FMR-1 gene and characterization of the fragile X mutation. Chromosoma 101: 381–387. 10.1007/BF00582832
- Orr HT, Chung M, Banfi S, Kwiatkowski TJ, Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LPW, Zoghbi HY. 1993. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 4: 221–226. 10.1038/ng0793-221
- ↵Owada K, Ishikawa K, Toru S, Ishida G, Gomyoda M, Tao O, Noguchi Y, Kitamura K, Kondo I, Noguchi E, 2005. A clinical, genetic, and neuropathologic study in a family with 16q-linked ADCA type III. Neurology 65: 629–632. 10.1212/01.wnl.0000173065.75680.e2
- ↵Pagnamenta AT, Kaiyrzhanov R, Zou Y, Da'as SI, Maroofian R, Donkervoort S, Dominik N, Lauffer M, Ferla MP, Orioli A, 2021. An ancestral 10-bp repeat expansion in VWA1 causes recessive hereditary motor neuropathy. Brain 144: 584–600. 10.1093/brain/awaa420
- ↵Pal A, Kretner B, Abo-Rady M, Glaβ H, Dash BP, Naumann M, Japtok J, Kreiter N, Dhingra A, Heutink P, 2021. Concomitant gain and loss of function pathomechanisms in C9ORF72 amyotrophic lateral sclerosis. Life Sci Alliance 4: e202000764. 10.26508/lsa.202000764
- Parrish JE, Oostra BA, Verkerk AJMH, Richards CS, Reynolds J, Spikes AS, Shaffer LG, Nelson DL. 1994. Isolation of a GCC repeat showing expansion in FRAXF, a fragile site distal to FRAXA and FRAXE. Nat Genet 8: 229–235. 10.1038/ng1194-229
- ↵Pearson CE. 2010. FSHD: a repeat contraction disease finally ready to expand (our understanding of its pathogenesis). PLoS Genet 6: e1001180. 10.1371/journal.pgen.1001180
- ↵Pearson CE, Edamura KN, Cleary JD. 2005. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet 6: 729–742. 10.1038/nrg1689
- Pembrey ME, Winter RM, Davies KE, Opitz JM, Reynolds JF. 1985. A premutation that generates a defect at crossing over explains the inheritance of fragile X mental retardation. Am J Med Genet 21: 709–717. 10.1002/ajmg.1320210413
- ↵Penrose LS. 1947. The problem of anticipation in pedigrees of dystrophia myotonica. Ann Eugen 14: 125–132. 10.1111/j.1469-1809.1947.tb02384.x
- ↵Pieretti M, Zhang F, Fu Y-H, Warren ST, Oostra BA, Caskey CT, Nelson DL. 1991. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66: 817–822. 10.1016/0092-8674(91)90125-I
- ↵Piras IS, Picinelli C, Iennaco R, Baccarin M, Castronovo P, Tomaiuolo P, Cucinotta F, Ricciardello A, Turriziani L, Nanetti L, 2020. Huntingtin gene CAG repeat size affects autism risk: family-based and case–control association study. Am J Med Genet 183: 341–351. 10.1002/ajmg.b.32806
- ↵Press MO, McCoy RC, Hall AN, Akey JM, Queitsch C. 2018. Massive variation of short tandem repeats with functional consequences across strains of Arabidopsis thaliana. Genome Res 28: 1169–1178. 10.1101/gr.231753.117
- ↵Pulst S-M, Nechiporuk A, Starkman S. 1993. Anticipation in spinocerebellar ataxia type 2. Nat Genet 5: 8–10. 10.1038/ng0993-8c
- Pulst S-M, Nechiporuk A, Nechiporuk T, Gispert S, Chen X-N, Lopes-Cendes I, Pearlman S, Starkman S, Orozco-Diaz G, Lunkes A, 1996. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 14: 269–276. 10.1038/ng1196-269
- ↵Quilez J, Guilmatre A, Garg P, Highnam G, Gymrek M, Erlich Y, Joshi RS, Mittelman D, Sharp AJ. 2016. Polymorphic tandem repeats within gene promoters act as modifiers of gene expression and DNA methylation in humans. Nucleic Acids Res 44: 3750–3762. 10.1093/nar/gkw219
- ↵Rafehi H, Szmulewicz DJ, Bennett MF, Sobreira NLM, Pope K, Smith KR, Gillies G, Diakumis P, Dolzhenko E, Eberle MA, 2019. Bioinformatics-based identification of expanded repeats: a non-reference intronic pentamer expansion in RFC1 causes CANVAS. Am J Hum Genet 105: 151–165. 10.1016/j.ajhg.2019.05.016
- ↵Ranum LPW, Cooper TA. 2006. RNA-mediated neuromuscular disorders. Annu Rev Neurosci 29: 259–277. 10.1146/annurev.neuro.29.051605.113014
- ↵Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72: 257–268. 10.1016/j.neuron.2011.09.010
- ↵Rhoads A, Au KF. 2015. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 13: 278–289. 10.1016/j.gpb.2015.08.002
- ↵Ricker K, Koch MC, Lehmann-Horn F, Pongratz D, Otto M, Heine R, Moxley RT. 1994. Proximal myotonic myopathy: a new dominant disorder with myotonia, muscle weakness, and cataracts. Neurology 44: 1448–1448. 10.1212/WNL.44.8.1448
- ↵Rio M, Malan V, Boissel S, Toutain A, Royer G, Gobin S, Morichon-Delvallez N, Turleau C, Bonnefont J-P, Munnich A, 2010. Familial interstitial Xq27.3q28 duplication encompassing the FMR1 gene but not the MECP2 gene causes a new syndromic mental retardation condition. Eur J Hum Genet 18: 285–290. 10.1038/ejhg.2009.159
- Ritchie RJ, Knight SJL, Hirst MC, Grewal PK, Bobrow M, Cross GS, Davies KE. 1994. The cloning of FRAXF: trinucleotide repeat expansion and methylation at a third fragile site in distal Xqter. Hum Mol Genet 3: 2115–2121. 10.1093/hmg/3.12.2115
- ↵Rodriguez CM, Todd PK. 2019. New pathologic mechanisms in nucleotide repeat expansion disorders. Neurobiol Dis 130: 104515. 10.1016/j.nbd.2019.104515
- ↵Rodriguez-Revenga L, Madrigal I, Pagonabarraga J, Xunclà M, Badenas C, Kulisevsky J, Gomez B, Milà M. 2009. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur J Hum Genet 17: 1359–1362. 10.1038/ejhg.2009.51
- ↵Rosas I, Martínez C, Clarimón J, Lleó A, Illán-Gala I, Dols-Icardo O, Borroni B, Almeida MR, van der Zee J, Van Broeckhoven C, 2020. Role for ATXN1, ATXN2, and HTT intermediate repeats in frontotemporal dementia and Alzheimer's disease. Neurobiol Aging 87: 139.e1–139.e7. 10.1016/j.neurobiolaging.2019.10.017
- ↵Ross CA, McInnis MG, Margolis RL, Li S-H. 1993. Genes with triplet repeats: candidate mediators of neuropsychiatric disorders. Trends Neurosci 16: 254–260. 10.1016/0166-2236(93)90175-L
- ↵Ruggieri A, Naumenko S, Smith MA, Iannibelli E, Blasevich F, Bragato C, Gibertini S, Barton K, Vorgerd M, Marcus K, 2020. Multiomic elucidation of a coding 99-mer repeat-expansion skeletal muscle disease. Acta Neuropathol 140: 231–235. 10.1007/s00401-020-02164-4
- ↵Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, Wakisaka A, Tashiro K, Ishida Y, Ikeuchi T, 1996. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 14: 277–284. 10.1038/ng1196-277
- ↵Santoro M, Masciullo M, Pietrobono R, Conte G, Modoni A, Bianchi MLE, Rizzo V, Pomponi MG, Tasca G, Neri G, 2013. Molecular, clinical, and muscle studies in myotonic dystrophy type 1 (DM1) associated with novel variant CCG expansions. J Neurol 260: 1245–1257. 10.1007/s00415-012-6779-9
- ↵Santoro M, Masciullo M, Silvestri G, Novelli G, Botta A. 2017. Myotonic dystrophy type 1: role of CCG, CTC and CGG interruptions within DMPK alleles in the pathogenesis and molecular diagnosis: variant interruptions in pathogenesis and molecular diagnosis of DM1. Clin Genet 92: 355–364. 10.1111/cge.12954
- Sarafidou T, Kahl C, Martinez-Garay I, Mangelsdorf M, Gesk S, Baker E, Kokkinaki M, Talley P, Maltby EL, French L, 2004. Folate-sensitive fragile site FRA10A is due to an expansion of a CGG repeat in a novel gene, FRA10AC1, encoding a nuclear protein. Genomics 84: 69–81. 10.1016/j.ygeno.2003.12.017
- ↵Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK, Smith DL, Faull RL, Roos RA, Howland D, 2013. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci 110: 2366–2370. 10.1073/pnas.1221891110
- ↵Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, Takahashi M, Matsuura T, Flanigan KM, Iwasaki S, 2009. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet 85: 544–557. 10.1016/j.ajhg.2009.09.019
- ↵Satoyoshi E, Kinoshita M. 1977. Oculopharyngodistal myopathy: report of four families. Arch Neurol 34: 89–92. 10.1001/archneur.1977.00500140043007
- ↵Savelyeva L, Brueckner LM. 2014. Molecular characterization of common fragile sites as a strategy to discover cancer susceptibility genes. Cell Mol Life Sci 71: 4561–4575. 10.1007/s00018-014-1723-z
- ↵Schalling M, Hudson TJ, Buetow KH, Housman DE. 1993. Direct detection of novel expanded trinucleotide repeats in the human genome. Nat Genet 4: 135–139. 10.1038/ng0693-135
- ↵Schneider A, Winarni TI, Cabal-Herrera AM, Bacalman S, Gane L, Hagerman P, Tassone F, Hagerman R. 2020. Elevated FMR1-mRNA and lowered FMRP – a double-hit mechanism for psychiatric features in men with FMR1 premutations. Transl Psychiatry 10: 205. 10.1038/s41398-020-00863-w
- ↵Scior A, Preissler S, Koch M, Deuerling E. 2011. Directed PCR-free engineering of highly repetitive DNA sequences. BMC Biotechnol 11: 87. 10.1186/1472-6750-11-87
- ↵Seixas AI, Loureiro JR, Costa C, Ordóñez-Ugalde A, Marcelino H, Oliveira CL, Loureiro JL, Dhingra A, Brandão E, Cruz VT, 2017. A pentanucleotide ATTTC repeat insertion in the non-coding region of DAB1, mapping to SCA37, causes spinocerebellar ataxia. Am J Hum Genet 101: 87–103. 10.1016/j.ajhg.2017.06.007
- ↵Sherman SL. 2000. Premature ovarian failure in the fragile X syndrome. Am J Med Genet 97: 189–194. 10.1002/1096-8628(200023)97:3<189::AID-AJMG1036>3.0.CO;2-J
- ↵Sherman SL, Morton NE, Jacobs PA, Turner G. 1984. The marker (X) syndrome: a cytogenetic and genetic analysis. Ann Human Genet 48: 21–37. 10.1111/j.1469-1809.1984.tb00830.x
- ↵Sherman SL, Jacobs PA, Morton NE, Froster-Iskenius U, Howard-Peebles PN, Nielsen KB, Partington MW, Sutherland GR, Turner G, Watson M. 1985. Further segregation analysis of the fragile X syndrome with special reference to transmitting males. Hum Genet 69: 289–299. 10.1007/BF00291644
- ↵Sherman RM, Forman J, Antonescu V, Puiu D, Daya M, Rafaels N, Boorgula MP, Chavan S, Vergara C, Ortega VE, 2019. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat Genet 51: 30–35. 10.1038/s41588-018-0273-y
- Shirazi SK, Bober MA, Coetzee GA. 1998. Polymorphic exonic CAG microsatellites in the gene amplified in breast cancer (AIB1 gene). Clin Genet 54: 102–103. 10.1111/j.1399-0004.1998.tb03704.x
- ↵Shortt JA, Ruggiero RP, Cox C, Wacholder AC, Pollock DD. 2020. Finding and extending ancient simple sequence repeat-derived regions in the human genome. Mob DNA 11: 11. 10.1186/s13100-020-00206-y
- Sisodia SS. 1998. Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell 95: 1–4. 10.1016/s0092-8674(00)81743-2
- ↵Snell RG, MacMillan JC, Cheadle JP, Fenton I, Lazarou LP, Davies P, MacDonald ME, Gusella JF, Harper PS, Shaw DJ. 1993. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington's disease. Nat Genet 4: 393–397. 10.1038/ng0893-393
- ↵Sone J, Mori K, Inagaki T, Katsumata R, Takagi S, Yokoi S, Araki K, Kato T, Nakamura T, Koike H, 2016. Clinicopathological features of adult-onset neuronal intranuclear inclusion disease. Brain 139: 3170–3186. 10.1093/brain/aww249
- ↵Sone J, Mitsuhashi S, Fujita A, Mizuguchi T, Hamanaka K, Mori K, Koike H, Hashiguchi A, Takashima H, Sugiyama H, 2019. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet 51: 1215–1221. 10.1038/s41588-019-0459-y
- ↵Song JHT, Lowe CB, Kingsley DM. 2018. Characterization of a human-specific tandem repeat associated with bipolar disorder and schizophrenia. Am J Hum Genet 103: 421–430. 10.1016/j.ajhg.2018.07.011
- ↵Striano P, Caranci F, Di Benedetto R, Tortora F, Zara F, Striano S. 2009. 1H-MR spectroscopy indicates prominent cerebellar dysfunction in benign adult familial myoclonic epilepsy. Epilepsia 50: 1491–1497. 10.1111/j.1528-1167.2008.01900.x
- Strømme P, Mangelsdorf ME, Shaw MA, Lower KM, Lewis SME, Bruyere H, Lütcherath V, Gedeon ÁK, Wallace RH, Scheffer IE, 2002. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet 30: 441–445. 10.1038/ng862
- ↵Summers K, Crespi B. 2008. The androgen receptor and prostate cancer: a role for sexual selection and sexual conflict? Med Hypotheses 70: 435–443. 10.1016/j.mehy.2007.04.044
- ↵Sureshkumar S, Todesco M, Schneeberger K, Harilal R, Balasubramanian S, Weigel D. 2009. A Genetic defect caused by a triplet repeat expansion in Arabidopsis thaliana. Science 323: 1060–1063. 10.1126/science.1164014
- ↵Sutherland GR. 1981. Heritable fragile sites on human chromosomes. VII. Children homozygous for the BrdU-requiring fra(10)(q25) are phenotypically normal. Am J Hum Genet 33: 946–949.
- ↵Sutherland GR, Baker E, Fratini A, Opitz JM, Reynolds JF. 1985. Excess thymidine induces folate sensitve fragile sites. Am J Med Genet 22: 433–443. 10.1002/ajmg.1320220234
- ↵Sutherland GR, Kremer E, Lynch M, Pritchard M, Yu S, Richards RI, Haan EA. 1991. Hereditary unstable DNA: a new explanation for some old genetic questions? Lancet 338: 289–292. 10.1016/0140-6736(91)90426-P
- ↵Sznajder ŁJ, Thomas JD, Carrell EM, Reid T, McFarland KN, Cleary JD, Oliveira R, Nutter CA, Bhatt K, Sobczak K, 2018. Intron retention induced by microsatellite expansions as a disease biomarker. Proc Natl Acad Sci 115: 4234–4239. 10.1073/pnas.1716617115
- ↵Tabib A, Vishwanathan S, Seleznev A, McKeown PC, Downing T, Dent C, Sanchez-Bermejo E, Colling L, Spillane C, Balasubramanian S. 2016. A polynucleotide repeat expansion causing temperature-sensitivity persists in wild Irish accessions of Arabidopsis thaliana. Front Plant Sci 7: 1311. 10.3389/fpls.2016.01311
- ↵Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA, Blair NF, Craufurd D, Priller J, Rickards H, 2019. Targeting Huntingtin expression in patients with Huntington's disease. N Engl J Med 380: 2307–2316. 10.1056/NEJMoa1900907
- ↵Tábuas-Pereira M, Almendra L, Almeida MR, Durães J, Pinho A, Matos A, Negrão L, Geraldo A, Santana I. 2019. Increased risk of melanoma in C9ORF72 repeat expansion carriers: a case–control study. Muscle Nerve 59: 362–365. 10.1002/mus.26383
- ↵Tang H, Kirkness EF, Lippert C, Biggs WH, Fabani M, Guzman E, Ramakrishnan S, Lavrenko V, Kakaradov B, Hou C, 2017. Profiling of short-tandem-repeat disease alleles in 12,632 human whole genomes. Am J Hum Genet 101: 700–715. 10.1016/j.ajhg.2017.09.013
- ↵Tankard RM, Bennett MF, Degorski P, Delatycki MB, Lockhart PJ, Bahlo M. 2018. Detecting expansions of tandem repeats in cohorts sequenced with short-read sequencing data. Am J Hum Genet 103: 858–873. 10.1016/j.ajhg.2018.10.015
- ↵Tezenas du Montcel S, Durr A, Bauer P, Figueroa KP, Ichikawa Y, Brussino A, Forlani S, Rakowicz M, Schöls L, Mariotti C, 2014. Modulation of the age at onset in spinocerebellar ataxia by CAG tracts in various genes. Brain 137: 2444–2455. 10.1093/brain/awu174
- ↵Tian Y, Wang J-L, Huang W, Zeng S, Jiao B, Liu Z, Chen Z, Li Y, Wang Y, Min H-X, 2019. Expansion of human-specific GGC repeat in neuronal intranuclear inclusion disease-related disorders. Am J Hum Genet 105: 166–176. 10.1016/j.ajhg.2019.05.013
- ↵Tilley WD, Marcelli M, Wilson JD, McPhaul MJ. 1989. Characterization and expression of a cDNA encoding the human androgen receptor. Proc Natl Acad Sci 86: 327–331. 10.1073/pnas.86.1.327
- ↵Tomé S, Holt I, Edelmann W, Morris GE, Munnich A, Pearson CE, Gourdon G. 2009. MSH2 ATPase domain mutation affects CTG•CAG repeat instability in transgenic mice. PLoS Genet 5: e1000482. 10.1371/journal.pgen.1000482
- ↵Tomé S, Dandelot E, Dogan C, Bertrand A, Geneviève D, Péréon Y, DM contraction study group, Simon M, Bonnefont J-P, Bassez G, 2018. Unusual association of a unique CAG interruption in 5′ of DM1 CTG repeats with intergenerational contractions and low somatic mosaicism. Hum Mutat 39: 970–982. 10.1002/humu.23531
- ↵Trost B, Engchuan W, Nguyen CM, Thiruvahindrapuram B, Dolzhenko E, Backstrom I, Mirceta M, Mojarad BA, Yin Y, Dov A, 2020. Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature 586: 80–86. 10.1038/s41586-020-2579-z
- ↵Trottier Y, Biancalana V, Mandel JL. 1994. Instability of CAG repeats in Huntington's disease: relation to parental transmission and age of onset. J Med Genet 31: 377–382. 10.1136/jmg.31.5.377
- ↵Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L, 1995. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature 378: 403–406. 10.1038/378403a0
- ↵Ummat A, Bashir A. 2014. Resolving complex tandem repeats with long reads. Bioinformatics 30: 3491–3498. 10.1093/bioinformatics/btu437
- ↵van der Sanden BPGH, Corominas J, de Groot M, Pennings M, Meijer RPP, Verbeek N, van de Warrenburg B, Schouten M, Yntema HG, Vissers LELM, 2021. Systematic analysis of short tandem repeats in 38,095 exomes provides an additional diagnostic yield. Genet Med 23: 1569–1573. 10.1038/s41436-021-01174-1
- van Deutekom JC, Wijmenga C, van Tienhoven EA, Gruter AM, Hewitt JE, Padberg GW, van Ommen GJ, Hofker MH, Frants RR. 1993. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet 2: 2037–2042. 10.1093/hmg/2.12.2037
- ↵van Dijk EL, Auger H, Jaszczyszyn Y, Thermes C. 2014. Ten years of next-generation sequencing technology. Trends Genet 30: 418–426. 10.1016/j.tig.2014.07.001
- ↵van Kuilenburg ABP, Tarailo-Graovac M, Richmond PA, Drögemöller BI, Pouladi MA, Leen R, Brand-Arzamendi K, Dobritzsch D, Dolzhenko E, Eberle MA, 2019. Glutaminase deficiency caused by short tandem repeat expansion in GLS. N Engl J Med 380: 1433–1441. 10.1056/NEJMoa1806627
- ↵Van Mossevelde S, van der Zee J, Cruts M, Van Broeckhoven C. 2017. Relationship between C9orf72 repeat size and clinical phenotype. Curr Opin Genet Dev 44: 117–124. 10.1016/j.gde.2017.02.008
- ↵van Wietmarschen N, Sridharan S, Nathan WJ, Tubbs A, Chan EM, Callen E, Wu W, Belinky F, Tripathi V, Wong N, 2020. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 586: 292–298. 10.1038/s41586-020-2769-8
- ↵Verkerk AJMH, Pieretti M, Sutcliffe JS, Fu Y-H, Kuhl DPA, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang F, 1991. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65: 905–914. 10.1016/0092-8674(91)90397-H
- ↵Vincent A, Hertz D, Petit C, Kretz C, Oberlé I, Mandel J-L. 1991. Abnormal pattern detected in fragile-X patients by pulsed-field gel electrophoresis. Nature 349: 624–626. 10.1038/349624a0
- ↵Virtaneva K, D'Amato E, Miao J, Koskiniemi M, Norio R, Avanzini G, Franceschetti S, Michelucci R, Tassinari CA, Omer S, 1997. Unstable minisatellite expansion causing recessively inherited myoclonus epilepsy, EPM1. Nat Genet 15: 393–396. 10.1038/ng0497-393
- ↵von Klopmann T, Ahonen S, Espadas-Santiuste I, Matiasek K, Sanchez-Masian D, Rupp S, Vandenberghe H, Rose J, Wang T, Wang P, 2021. Canine Lafora disease: an unstable repeat expansion disorder. Life 11: 689. 10.3390/life11070689
- ↵Wang M-D, Gomes J, Cashman NR, Little J, Krewski D. 2014. Intermediate CAG repeat expansion in the ATXN2 gene is a unique genetic risk factor for ALS−a systematic review and meta-analysis of observational studies. PLoS One 9: e105534. 10.1371/journal.pone.0105534
- ↵Warren ST, Zhang F, Licameli GR, Peters JF. 1987. The fragile X site in somatic cell hybrids: an approach for molecular cloning of fragile sites. Science 237: 420–423. 10.1126/science.3603029
- ↵Webb AA, McMillan C, Cullen CL, Boston SE, Turnbull J, Minassian BA. 2009. Lafora disease as a cause of visually exacerbated myoclonic attacks in a dog. Can Vet J 50: 963–967.
- ↵Weissensteiner MH, Pang AWC, Bunikis I, Höijer I, Vinnere-Petterson O, Suh A, Wolf JBW. 2017. Combination of short-read, long-read, and optical mapping assemblies reveals large-scale tandem repeat arrays with population genetic implications. Genome Res 27: 697–708. 10.1101/gr.215095.116
- ↵Wenger AM, Peluso P, Rowell WJ, Chang P-C, Hall RJ, Concepcion GT, Ebler J, Fungtammasan A, Kolesnikov A, Olson ND, 2019. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol 37: 1155–1162. 10.1038/s41587-019-0217-9
- ↵Wieben ED, Aleff RA, Tosakulwong N, Butz ML, Highsmith WE, Edwards AO, Baratz KH. 2012. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts fuchs corneal dystrophy. PLoS One 7: e49083. 10.1371/journal.pone.0049083
- Wijmenga C, Hewitt JE, Sandkuijl LA, Clark LN, Wright TJ, Dauwerse HG, Gruter A-M, Hofker MH, Moerer P, Williamson R, 1992. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet 2: 26–30. 10.1038/ng0992-26
- ↵Winnepenninckx B, Debacker K, Ramsay J, Smeets D, Smits A, FitzPatrick DR, Kooy RF. 2007. CGG-Repeat expansion in the DIP2B gene is associated with the fragile site FRA12A on chromosome 12q13.1. Am J Hum Genet 80: 221–231. 10.1086/510800
- ↵Wöhrle D, Kotzot D, Hirst MC, Manca A, Korn B, Schmidt A, Barbi G, Rott HD, Poustka A, Davies KE. 1992. A microdeletion of less than 250 kb, including the proximal part of the FMR-I gene and the fragile-X site, in a male with the clinical phenotype of fragile-X syndrome. Am J Hum Genet 51: 299–306.
- ↵Wright GEB, Collins JA, Kay C, McDonald C, Dolzhenko E, Xia Q, Bečanović K, Drögemöller BI, Semaka A, Nguyen CM, 2019. Length of uninterrupted CAG, independent of polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am J Hum Genet 104: 1116–1126. 10.1016/j.ajhg.2019.04.007
- ↵Xu G-L, Bestor TH, Bourc'his D, Hsieh C-L, Tommerup N, Bugge M, Hulten M, Qu X, Russo JJ, Viegas-Péquignot E. 1999. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 402: 187–191. 10.1038/46052
- ↵Yeetong P, Pongpanich M, Srichomthong C, Assawapitaksakul A, Shotelersuk V, Tantirukdham N, Chunharas C, Suphapeetiporn K, Shotelersuk V. 2019. TTTCA repeat insertions in an intron of YEATS2 in benign adult familial myoclonic epilepsy type 4. Brain 142: 3360–3366. 10.1093/brain/awz267
- ↵Yeetong P, Chunharas C, Pongpanich M, Bennett MF, Srichomthong C, Pasutharnchat N, Suphapeetiporn K, Bahlo M, Shotelersuk V. 2021. Founder effect of the TTTCA repeat insertions in SAMD12 causing BAFME1. Eur J Hum Genet 29: 343–348. 10.1038/s41431-020-00729-1
- ↵Yu S, Pritchard M, Kremer E, Lynch M, Nancarrow J, Baker E, Holman K, Mulley JC, Warren ST, Schlessinger D, 1991. Fragile X genotype characterized by an unstable region of DNA. Science 252: 1179–1181. 10.1126/science.252.5009.1179
- ↵Yu S, Mangelsdorf M, Hewett D, Hobson L, Baker E, Eyre HJ, Lapsys N, Le Paslier D, Doggett NA, Sutherland GR, 1997. Human chromosomal fragile site FRA16B is an amplified AT-rich minisatellite repeat. Cell 88: 367–374. 10.1016/S0092-8674(00)81875-9
- ↵Yu Z, Zhu Y, Chen-Plotkin AS, Clay-Falcone D, McCluskey L, Elman L, Kalb RG, Trojanowski JQ, Lee VM-Y, Van Deerlin VM, 2011. PolyQ repeat expansions in ATXN2 associated with ALS Are CAA interrupted repeats. PLoS One 6: e17951. 10.1371/journal.pone.0017951
- Yu J, Deng J, Guo X, Shan J, Luan X, Cao L, Zhao J, Yu M, Zhang W, Lv H, 2021. The GGC repeat expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy type 3. Brain 144: 1819–1832. 10.1093/brain/awab077
- Yuan Y, Liu Z, Hou X, Li W, Ni J, Huang L, Hu Y, Liu P, Hou X, Xue J, 2020. Identification of GGC repeat expansion in the NOTCH2NLC gene in amyotrophic lateral sclerosis. Neurology 95: e3394–e3405. 10.1212/WNL.0000000000010945
- ↵Zambonin JL, Bellomo A, Ben-Pazi H, Everman DB, Frazer LM, Geraghty MT, Harper AD, Jones JR, Kamien B, Kernohan K, 2017. Spinocerebellar ataxia type 29 due to mutations in ITPR1: a case series and review of this emerging congenital ataxia. Orphanet J Rare Dis 12: 121. 10.1186/s13023-017-0672-7
- ↵Zhao X, Su L, Schaack S, Sadd BM, Sun C. 2018. Tandem repeats contribute to coding sequence variation in bumblebees (Hymenoptera: Apidae). Genome Biol Evol 10: 3176–3187. 10.1093/gbe/evy244
- Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC. 1997. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A-voltage-dependent calcium channel. Nat Genet 15: 62–69. 10.1038/ng0197-62
- ↵Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, 2011. Non-ATG–initiated translation directed by microsatellite expansions. Proc Natl Acad Sci 108: 260–265. 10.1073/pnas.1013343108