Abstract
Establishing the molecular basis of DNA mutations that cause inherited disease is of fundamental importance to understanding the origin, nature, and clinical sequelae of genetic disorders in humans. The majority of disease-associated mutations constitute single-base substitutions and short deletions and/or insertions resulting from DNA replication errors and the repair of damaged bases. However, pathological mutations can also be introduced by nonreciprocal recombination events between paralogous sequences, a phenomenon known as interlocus gene conversion (IGC). IGC events have thus far been linked to pathology in more than 20 human genes. However, the large number of duplicated gene sequences in the human genome implies that many more disease-associated mutations could originate via IGC. Here, we have used a genome-wide computational approach to identify disease-associated mutations derived from IGC events. Our approach revealed hundreds of known pathological mutations that could have been caused by IGC. Further, we identified several dozen high-confidence cases of inherited disease mutations resulting from IGC in ∼1% of all genes analyzed. About half of the donor sequences associated with such mutations are functional paralogous genes, suggesting that epistatic interactions or differential expression patterns will determine the impact upon fitness of specific substitutions between duplicated genes. In addition, we identified thousands of hitherto undescribed and potentially deleterious mutations that could arise via IGC. Our findings reveal the extent of the impact of interlocus gene conversion upon the spectrum of human inherited disease.
The number of mutations responsible for—or associated with—human inherited disease has grown significantly over the past three decades, and now exceeds 117,000 different DNA lesions in >4300 genes (Stenson et al. 2009b; Cooper et al. 2010). Studying the molecular processes responsible for introducing pathological mutations into the human genome is of primary importance with respect to improving our understanding of the nature, onset, and course of mendelian disease. DNA sequence analysis has demonstrated that most of the known pathological mutations are small-scale lesions involving single nucleotides or short (≤20 bp) deletions/insertions of DNA within coding regions, regulatory sequences, or splice sites, suggesting that DNA replication and DNA repair are the main pathways acting in the genesis of pathological mutations (Kondrashov and Rogozin 2004; Ball et al. 2005; Cooper et al. 2010, 2011). In addition, though they are often much harder to detect, mutational processes including nonhomologous end-joining (NHEJ), fork stalling and template switching (FoSTeS), nonallelic homologous recombination, and retrotransposon insertion are responsible for gross genomic rearrangements events that have been linked to human inherited disease (Cooper et al. 2011). For example, NHEJ is responsible for the most common type of Robertsonian translocation between chromosomes 11 and 22 (Kurahashi et al. 2010); FoSTeS has been implicated in the onset of the Pelizaeus-Merzbacher disease, an X-linked dysmyelinating disorder (Lee et al. 2007); nonallelic homologous recombination is known to cause several disease-associated mutations, such as microdeletions in the gene NF1 that lead to neurofibromatosis type 1 (Lopez-Correa et al. 2001); finally, insertions of L1, Alu, and SVA retrotransposons have been implicated in mendelian disease since the discovery of a de novo L1 element integration causing Hemophilia A in 1988 (Kazazian et al. 1988). Single-nucleotide mutations can originate through the action of several processes, including the incorporation of a noncomplementary nucleotide during DNA replication or the chemical modification of a base that serves to increase the rate of mutation, for instance, methylation of cytosine in a CpG dinucleotide leading to a C-to-T transition (Cooper and Krawczak 1993). Interestingly, most pathological microinsertions and microdeletions involve, respectively, the duplication and deletion of short DNA repeats, implicating DNA replication slippage as the main mechanism responsible for these lesions (Kondrashov and Rogozin 2004; Ball et al. 2005).
Pathogenic mutations can also be introduced by interlocus gene conversion (IGC) between disease-associated genes and their paralogous sequences. In these recombination events, genetic information is transferred from a donor locus to an acceptor locus (Arnheim et al. 1980; Miyata et al. 1980; Scherer and Davis 1980). When the acceptor sequence is a functional gene, IGC can introduce deleterious nucleotide changes into the new context. Recent literature surveys have identified a total of 30 unique, nonoverlapping mutations in 23 genes associated with human inherited disease that were caused by IGC (Chen et al. 2007; Chuzhanova et al. 2009). These disease mutations were templated by donor sequences that were either putatively nonfunctional gene copies (pseudogenes) or functional paralogous genes (Chen et al. 2007; Chuzhanova et al. 2009). Given that a significant proportion of the >21,000 annotated protein-coding genes in the human genome have multiple homologous sequences, either evolutionarily related protein-coding genes or nonfunctional pseudogenes (Torrents et al. 2003; Zhang and Gerstein 2004; Clamp et al. 2007), we reasoned that IGC could be responsible for a much higher number of disease-associated mutations than has hitherto been appreciated. In this study, we analyzed 60,488 different disease mutations in 3196 genes reported in the Human Gene Mutation Database (HGMD, Professional release 2010.3) (Stenson et al. 2009a) to disclose instances of IGC-derived disease alleles. Our findings indicate that IGC introduces disease-associated mutations in ∼1% of analyzed human genes. In addition, we predict thousands of potential IGC-mediated amino acid replacements with deleterious effects in almost 900 different human genes, 158 of which have already been implicated in human disease.
Results and Discussion
Pathological mutations derived from interlocus gene conversion
We first determined the proportion of genes with disease mutations listed in HGMD that have paralogous sequences capable, in principle, of mediating IGC. We found that a total of 441 genes with mutations reported in the HGMD database (hereafter termed HGMD genes) have at least one paralogous sequence elsewhere in the genome sharing ≥92% sequence identity over a ≥200-bp stretch. This level of sequence identity is thought to be needed to mediate interlocus gene conversion events (Waldman and Liskay 1988). The proportion of HGMD genes with paralogs (14%) is comparable to the proportion (17%) exhibited by 16,181 genes not present in the HGMD database (hereafter termed non-HGMD genes) with potential paralogous donor sequences. Of the 441 HGMD genes with at least one paralog, 60 have paralogs that would introduce known disease alleles (n = 168 unique mutations in HGMD) (Supplemental Table 1). These paralogous sequences harbor a large pool of potential disease alleles that could in principle be introduced into functional genes by IGC (cf. Bischof et al. 2006).
We then attempted to identify specific instances of inherited human gene mutations introduced by IGC. We initially identified 243 paralogous sequences that could act as donors of the above-mentioned 168 unique mutations found in HGMD. We consider these sequences to be “candidate” donors involved in the gene conversion events leading to the observed disease mutations in the acceptor genes (Table 1). Because the same allelic state could appear in parallel in both the disease gene and its paralog, strong evidence for the role of IGC requires the co-occurrence of multiple mutations in the disease allele, all perfectly matching the paralogous (donor) sequence (Chen et al. 2007). Therefore, to identify such “high-confidence candidates,” we required the known disease allele sequence to match perfectly the possible donor sequence at the site of the putative pathological mutation and, minimally, at one additional noncontiguous site that differed between the disease and nondisease alleles (Supplemental Fig. 1). We found 22 instances of disease alleles in 16 genes that were supported by co-occurring substitutions, nine of which had not previously been described in surveys of IGC events responsible for pathological mutations (Supplemental Tables 2, 3). We then combined our results with those from recent literature surveys that applied the same criteria to define high-confidence IGC events (Chen et al. 2007; Chuzhanova et al. 2009), and found that interlocus gene conversion is able to account for at least 41 disease alleles in 30 genes, corresponding to ∼1% of the 3196 human disease genes analyzed here (Table 1; Supplemental Table 4). Most of the gene lesions in these disease alleles are reported as disease-causing mutations in HGMD, although several have been classified as disease-associated or functional polymorphisms (Supplemental Table 4).
It is possible that some of these disease alleles could have originated from multiple mutations occurring independently, instead of via a recombination event. However, several lines of evidence strongly support interlocus gene conversion as the mechanism responsible for introducing the observed pathological mutations. First, the probability by chance alone of multiple mutations occurring not only at the specific sites that serve to distinguish the proposed donor and acceptor sequences, but also for these mutations to correspond to the exact same bases present in the donor sequences, is infinitesimally low. Although we required that at least two nucleotide substitutions should match between the disease allele and the donor sequence (without any mismatches), in practice, the majority of our high-confidence disease alleles (29/41) harbored between 3 and 22 nucleotide differences with respect to the wild-type allele (Supplemental Tables 2, 3; Chuzhanova et al. 2009), thereby further lowering the probability that such a disease allele could have arisen via independent mutations at multiple sites. Second, we found evidence of gene conversion between the donor and acceptor sequences in 37/41 cases of the high-confidence data set using GENECONV, a program specifically developed to detect gene conversion tracts in sequence alignments (Sawyer 1989). All four cases that GENECONV failed to detect comprised two mutations, while three were characterized by very short (38–56 bp) maximal conversion tracts. Statistically based methods such as the tests implemented in GENECONV have insufficient power to identify short conversion tracts that harbor few mutations. Third, the DNA sequences involved in 27 of the 41 high-confidence IGC events were enriched in recombination-inducing motifs and alternative non-B DNA conformations, both of which are known to promote gene conversion events (Chuzhanova et al. 2009). Fourth, at least 10 examples of high-confidence IGC have been described in family-based studies, where the disease allele has arisen de novo in a single generation, events which would be extremely unlikely to originate through multiple independent mutations. Fifth, although multiple mutations can arise independently through mechanisms other than gene conversion (Chen et al. 2009; Schrider et al. 2011), the component mutations of such complex lesions tend to be clustered along the chromosome with the majority occurring within 4 bp of one another (Schrider et al. 2011). Only four of the minimum observed tract lengths among our high-confidence set of disease alleles were <10 bp in length, providing further evidence to support our contention that gene conversion is the source of these disease mutations.
Donor sequences of IGC-derived pathological mutations
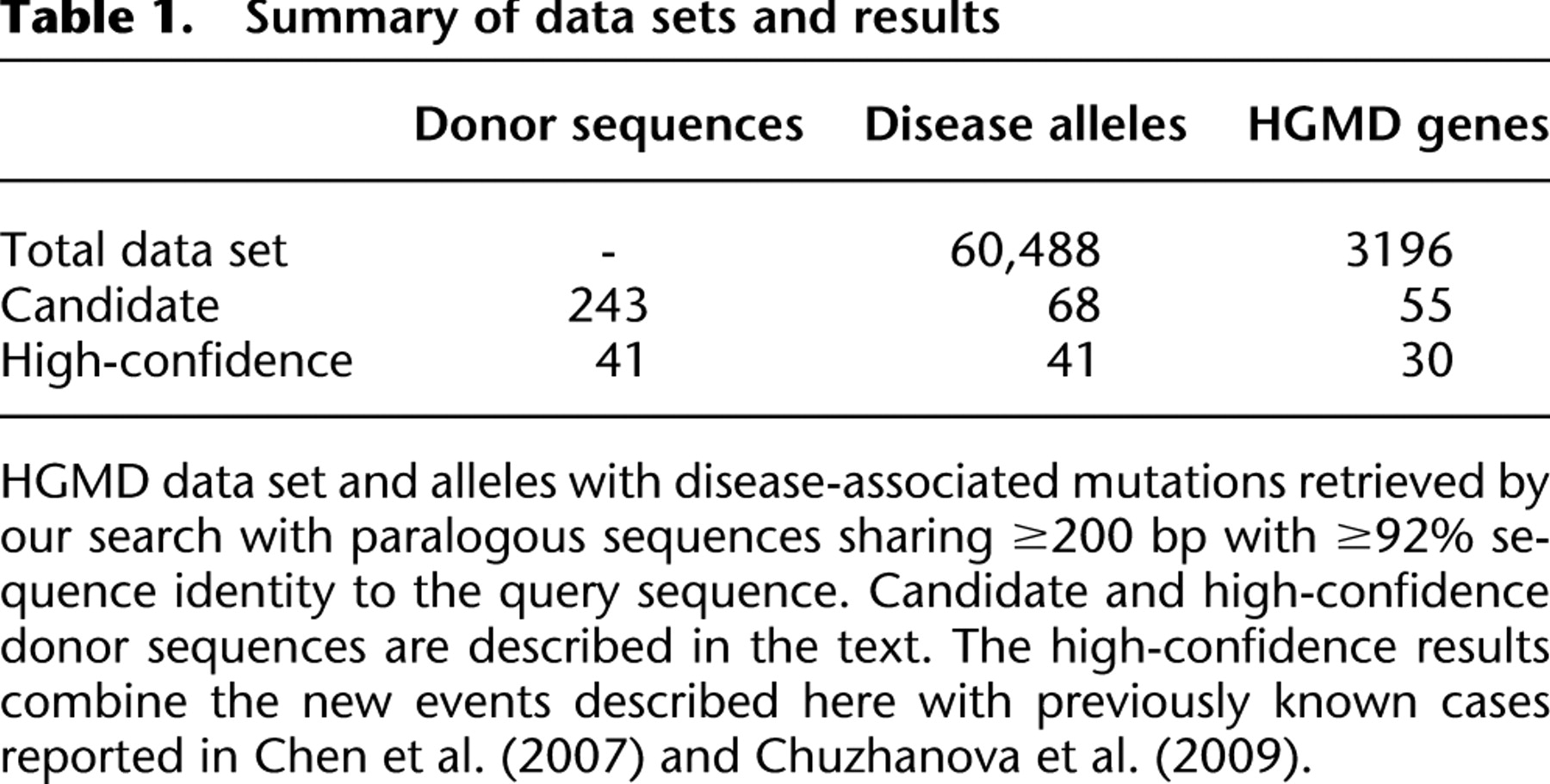
Of our high-confidence candidate donor sequences that appear capable of introducing disease alleles, approximately half are paralogous pseudogenes (Table 2). When acting as donor sequences in IGC events, pseudogenes generally introduce either missense or nonsense mutations, or indels that give rise to frameshifts. The vast majority of the pseudogene donor sequences were found to be nonprocessed copies, i.e., they were derived from a genomic DNA-mediated duplication of the acceptor gene (Jun et al. 2009). However, we also found evidence supporting the occurrence of IGC from a processed pseudogene to the functional gene GJA1, encoding the gap junction protein connexin 43, which was associated with hypoplastic left heart syndrome (Dasgupta et al. 2001). Processed pseudogenes originate when mRNA is reverse-transcribed into cDNA that is then subsequently integrated into the nuclear genome (Podlaha and Zhang 2009). We noted that multiple disease mutations in the GJA1 gene were introduced via gene conversion from a processed GJA1 pseudogene. Indeed, we detected two different regions within GJA1 disease alleles harboring two or more missense mutations that appear identical to the sequence of the GJA1 processed pseudogene, GJA1P1, which shares ∼96% nucleotide identity with GJA1 (Fig. 1). Two previous studies (Dasgupta et al. 2001; Chen et al. 2005) have reported disease states associated with these missense mutations, together with several silent mutations within the coding region of GJA1, all of which match the pseudogene sequence. The recent identification of many short processed pseudogenes in the human genome (Terai et al. 2010) suggests that pseudogenes may be even more common than previously thought. If such paralogous sequences were also capable of acting as templates for IGC events, then the impact of this phenomenon would almost certainly be greater than has previously been appreciated, particularly as those IGC events donating relatively short sequences are likely to be much less frequently recognized (i.e., they will almost invariably be considered to be single base-pair substitutions).
IGC donor sequences involved in high-confidence IGC events
Interlocus gene conversion (IGC) from the human processed pseudogene GJA1P1 to its functional paralog, GJA1. The chromosomal locations of GJA1 and GJA1P1 are 6q22.31 and 5q21.3, respectively. The filled black box in GJA1 represents the coding sequence, whereas the empty boxes denote the 5′UTR and 3′UTR. The light-gray shadings connect regions of similarity between the two sequences and serve to highlight the absence of intron 1 in the pseudogene. Regions representing the minimal conversion tract are connected by dark gray shading. The sequence in these regions is identical between the GJA1 disease allele and GJA1P1. A second, independent IGC event between the two paralogous sequences is not shown here.
Almost half of the donor sequences found to have introduced disease alleles were themselves functional genes (Table 2). Evidence for a functional effect in the majority of these cases (13/15) has been supported by experimental data, including reporter gene assays, in vitro assays of enzymatic activity, and analysis of gene expression variation due to changes in the regulatory regions of acceptor genes (Supplemental Table 5). In the case of coding sequence changes, it may be inferred that the amino acid changes that compromise the function of the protein encoded by the acceptor gene are not deleterious with respect to the function of the protein encoded by the donor gene. Further, these results imply that the donor gene proteins may have accumulated additional amino acid changes elsewhere within their sequences, which compensate for the presence of these conditionally deleterious mutations (Kondrashov et al. 2002). Alternatively, the protein encoded by the acceptor gene might be involved in specific protein–protein interactions that are negatively affected by the disease-related amino acid change; the protein encoded by the donor gene would not share these interactions if, for example, the two paralogous genes were to have different spatial or temporal expression patterns.
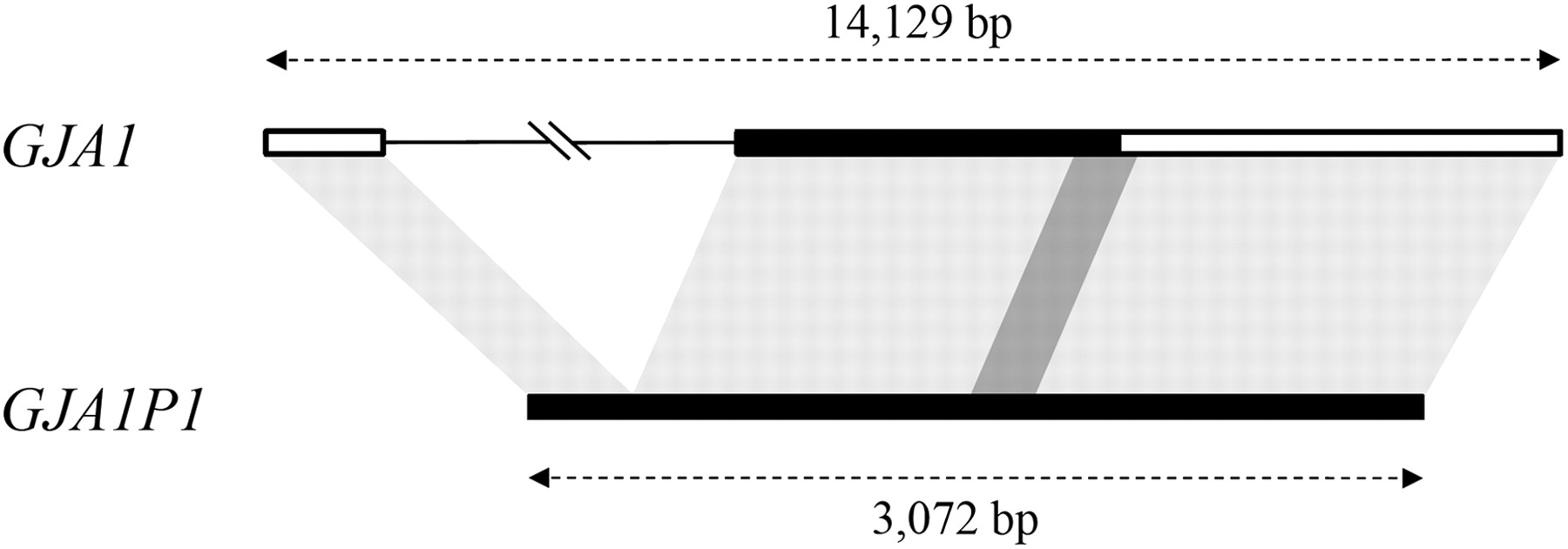
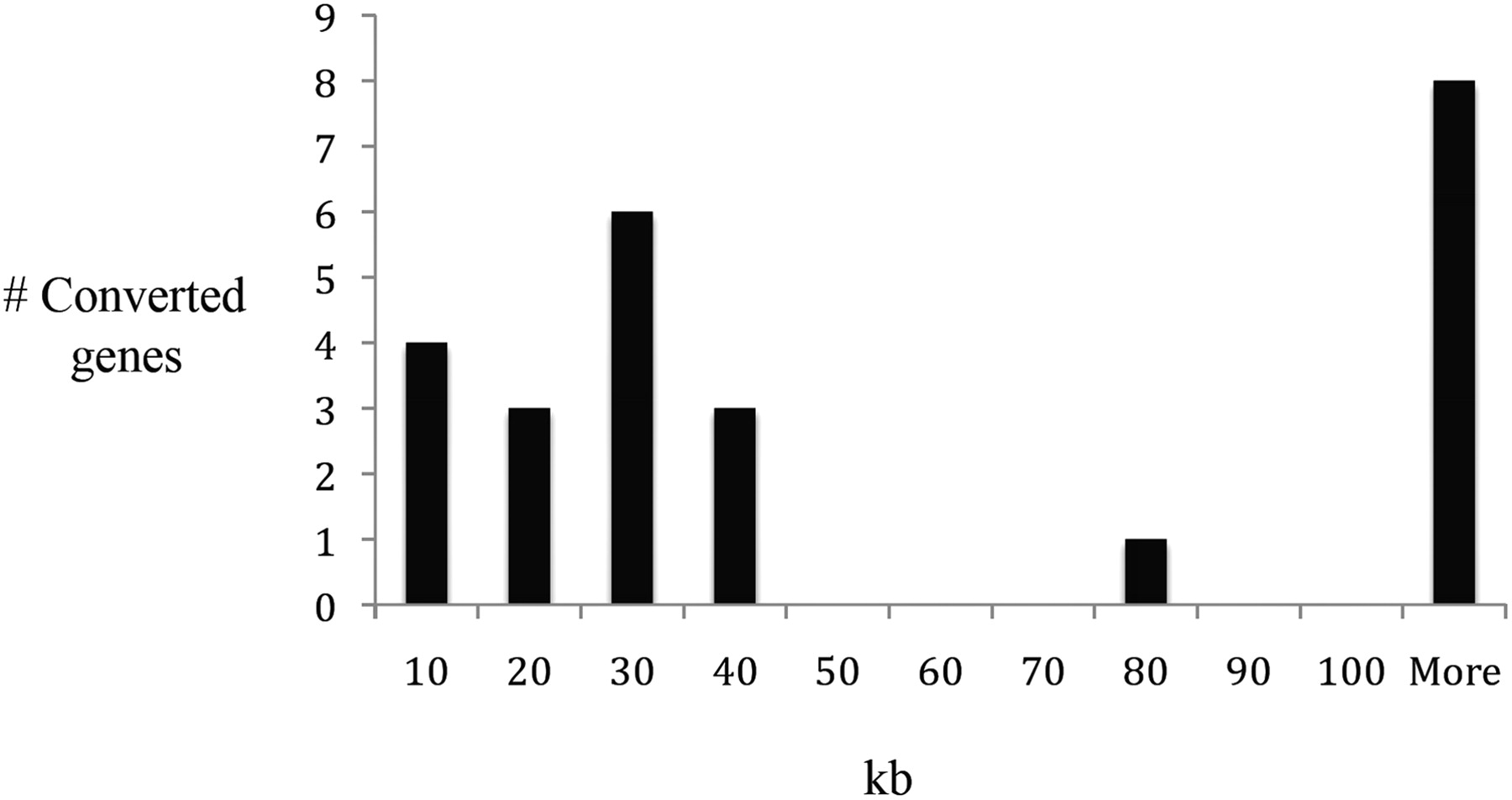
Ninety percent of documented IGC events were found to involve donor and acceptor sequences on the same chromosome, considerably higher than the proportions of HGMD genes (43%) and non-HGMD genes (52%) that have intrachromosomal paralogous sequences (Fig. 2; Supplemental Table 6). We also found that almost 70% of the intrachromosomal donor/acceptor IGC pairs are closely linked, indeed, <100 kb apart (Fig. 3). It has become apparent from previous genome-wide studies of paralogous genes (Ezawa et al. 2006; McGrath et al. 2009; Hsu et al. 2010) that both chromosomal distribution and physical linkage play a major role in IGC; the results presented here, derived from disease gene data, imply that chromosomal location also plays an important role in the likelihood of acquiring deleterious mutations via IGC.
Excess of interlocus gene conversion (IGC) between intrachromosomal paralogous sequences. HGMD genes with IGC: 30 genes with deleterious mutations introduced by IGC. All HGMD genes: 3196 genes from the HGMD database.
Distance in kilobases (kb) between donor and acceptor sequences for the 25 intrachromosomal IGC pairs with assigned donor sequences.
Our observations may help to explain the low rate of IGC between functional genes and processed pseudogenes, since the latter integrate virtually at random into the genome—and hence, are often found on a different chromosome from the parental gene. Because processed pseudogenes generally lack introns, sequence homology with their parent genes is limited to individual exons, which represents another factor that would militate against recombination involving this type of pseudogene. Consistent with this idea, both the coding sequence and 3′UTR of the GJA1 gene lack introns and, hence, would align across almost the entire sequence of GJA1P1; this unusual gene architecture may well have facilitated the IGC events we detected (Fig. 1). The importance of the chromosomal distribution and genomic architecture of paralogous gene/pseudogene sequences in IGC events is also illustrated by the over-representation of nonprocessed pseudogenes among donor sequences in our data set (Table 2), despite being about sixfold less abundant than processed pseudogenes. Indeed, 51% of nonprocessed pseudogenes are on the same chromosome as their parental genes, as compared with 13% of processed pseudogenes, and in most cases they share their parental genes' exon/intron structure.
Deleterious mutations potentially introduced by IGC
It has been proposed that known pathological mutations represent only a small fraction of all possible deleterious mutations in our species (Cooper et al. 2010). Under some circumstances, the effect of a specific mutation upon fitness can be evaluated given the possible structural and/or functional consequences for the affected gene and protein. For example, the potentially deleterious effect of an amino acid replacement can be predicted by taking advantage of data pertaining to the protein's secondary structure, the evolutionary conservation of orthologous protein sequences, and other structural features (Ramensky et al. 2002; Ng and Henikoff 2003).
Given the high number of paralogous gene sequences found in our own and previous studies, we reasoned that many yet-undescribed deleterious mutations responsible for nonsynonymous changes in functional genes could be mediated by IGC events. To ascertain the scale of this potential source of deleterious amino acid replacements, we first retrieved every protein site that could be changed by an IGC event templated by a donor sequence, including mutations leading to premature stop codons in the coding sequence of a gene. Second, we inferred the severity of such amino acid replacements using the PolyPhen-2 software package, which uses 11 features in order to predict the fitness impact of a given change in a protein's primary sequence and displays higher levels of accuracy and specificity than other available methods (Adzhubei et al. 2010).
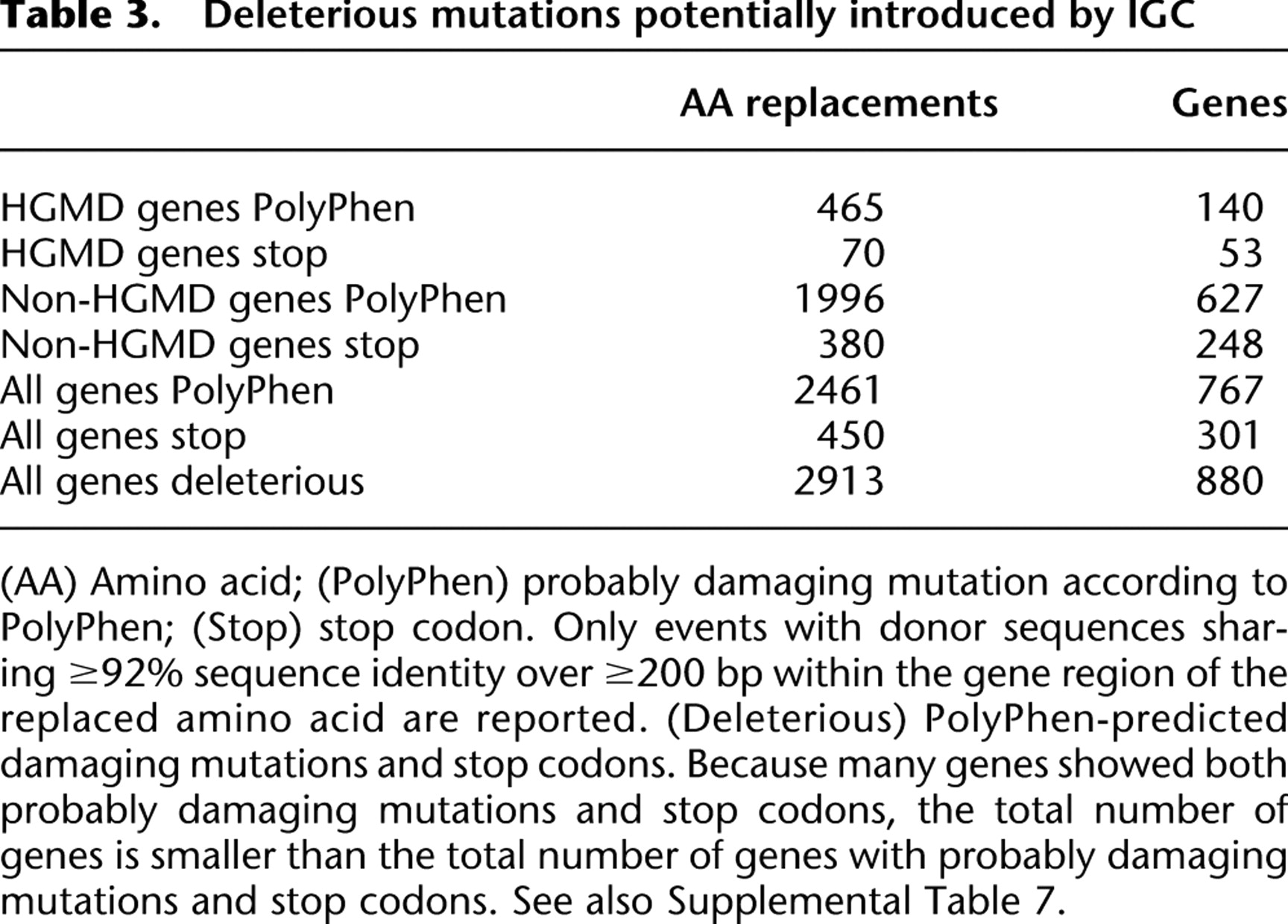
By BLASTing ∼19,500 human genes against the human genome, we identified more than 55,000 amino acid replacements classified by PolyPhen-2 as probably damaging, together with ∼8000 premature stop codons that could be introduced by IGC events (Supplemental Table 7). When we restricted our data set to probably damaging mutations from donor sequences sharing ≥92% identity with the acceptor sequence (Waldman and Liskay 1988; Chen et al. 2007; Wolf et al. 2009) over a stretch of ≥200 bp, we still obtained 2461 deleterious amino acid changes in 767 genes, and 450 premature stop codons in 301 genes (Table 3). A total of 19% of these mutations are potentially damaging to 158 HGMD genes (i.e., genes known to cause human disease when disrupted). In both HGMD and non-HGMD gene data sets, ∼80% of the possible donor sequences of such mutations are pseudogenes (Supplemental Table 8), a higher proportion than that evident with all possible paralogous sequences (Supplemental Table 6). This enrichment of pseudogenes as donor sequences of predicted deleterious mutations is likely to reflect the lack of selective pressure operating on nonfunctional coding sequences to remove harmful amino acid replacements. Consistent with this postulate, the 450 predicted premature stop codon mutations were found to be derived mainly from pseudogene donor sequences. However, we also detected 31 premature stop codons where the donor sequence was a functional gene, pointing to the dynamic evolution of the 3′UTR and exon structure between these paralogous genes (Fig. 4; Supplemental Table 9).
Deleterious mutations potentially introduced by IGC
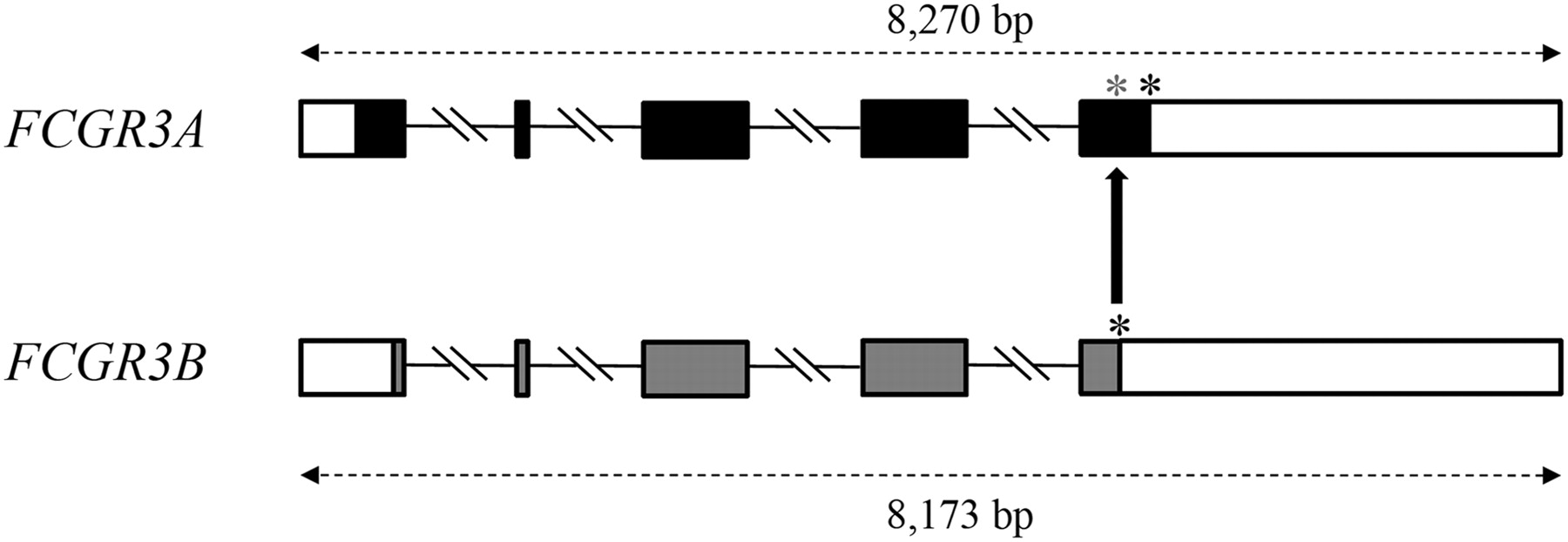
Interlocus gene conversion from FCGR3B to FCGR3A. The FCGR3A gene encodes a receptor for the Fc portion of immunoglobulin G, which is involved in the removal of antigen–antibody complexes from the circulation. The highly similar paralog, FCGR3B, which resides only 73 kb downstream from FCGR3A, encodes a shorter protein due to differences in the positions of both the start and stop codons. Coding exons are shown as filled boxes, UTRs as empty boxes. Introns are represented as lines and are not drawn to scale. The black asterisks indicate the normal stop codons of the FCGR3A and FCGR3B genes; the gray asterisk identifies the premature stop codon in the FCGR3A gene that is introduced by an IGC event with FCGR3B.
Some of the ∼3000 potential IGC-mediated deleterious mutations would affect genes that are known to experience pathological mutations due to IGC events (Supplemental Table 10). For example, seven amino acid changes, originating via IGC, are known to be responsible for autosomal dominant polycystic kidney disease type 1 through alteration of the sequence of the PKD1 gene; we identified 30 new and potentially deleterious amino acid replacements that could in principle be introduced by IGC between PKD1 and any one of its six nonprocessed pseudogenes on chromosome 16.
In addition, we identified hundreds of deleterious mutations that could potentially be introduced by IGC in disease-associated (HGMD) genes where gene conversion—at least to our knowledge—has not yet been implicated as a mechanism causing disease-associated lesions (Supplemental Table 11). In many such instances, the possible donor and acceptor sequences lie within a few million bases of each other (Supplemental Table 11), a configuration that facilitates gene conversion between paralogous sequences (Ezawa et al. 2006; Benovoy and Drouin 2009; McGrath et al. 2009). The RANBP2 gene is a striking example in this regard, with its seven paralogous genes on chromosome 2 separated by only ∼1–22 Mb. PolyPhen-2 predicted 24 deleterious mutations in RANBP2 that could originate from IGC events involving these paralogs as donor sequences.
Conclusions
The structural and functional redundancy of the human genome, and in particular its duplicated gene copies, provide the fuel for homologous recombination events that can cause inherited disease and cancer (Chen et al. 2010; Cooper et al. 2011). We have shown that interlocus gene conversion, a common consequence of homologous recombination, is responsible for at least 41 distinct disease alleles, but could also introduce many hundreds of other mutations that cause human inherited disease. Finally, we identified thousands of potentially deleterious amino acid replacements and premature stop codons in human genes that could, in principle, be introduced by IGC events, including 535 sites in 158 HGMD genes, suggesting that the role of interlocus gene conversion as a source of human pathological mutations could be much greater than hitherto appreciated.
Methods
Databases
Mutation data were retrieved from the HGMD Professional release 2010.3 (September 2010). Microlesions in the following categories were used for further analysis: missense/nonsense, indels, and regulatory single base-pair substitutions (Supplemental Table 4). Mutations due to microdeletions and microinsertions (≤20 bp) were not included, because such lesions could be due to other common mutational mechanisms, including slippage of the DNA polymerase during DNA replication (Kondrashov and Rogozin 2004; Ball et al. 2005). Indeed, in the case of 70% of microdeletions and 89% of microinsertions reported in HGMD, the exact position of the lesion cannot be unequivocally determined due to the repetitive nature of these microlesion sites. A total of 60,488 different pathological mutations were therefore used in this analysis.
Some known disease-related mutations due to IGC were not detected by our approach. In general, this was because they represented multiple base changes, which are recorded singly in HGMD as “complex lesions” rather than as their individual component missense/nonsense substitutions. In addition, and in accord with the inclusion criteria adopted in a previous survey (Chuzhanova et al. 2009), 23 IGC events from nine genes were excluded from our final data set because their maximal conversion tracts overlapped with other events reported here. The genome coordinates of those pseudogenes used to infer the proportion of nonprocessed and processed intrachromosomal pseudogene–gene pairs were retrieved from the human pseudogene database (www.pseudogene.org).
Sequence similarity searches
To retrieve all possible donor sequences of the documented IGC events, we performed sequence similarity searches using BLAST (Altschul et al. 1990), and used different stretches of 200 bp around each mutation reported in HGMD as query sequences (see below). From the paralogous sequences retrieved using this approach, we selected those sequences that shared ≥92% sequence identity (Waldman and Liskay 1988; Chen et al. 2007; Wolf et al. 2009) with the acceptor sequence over a stretch of ≥200 bp and which also contained the same mutation as the disease allele, for further investigation. Genome coordinates and sequence information available for each HGMD mutation were used to obtain three sets of genome sequences that we termed “around,” “upstream,” and “downstream”; these contained, respectively, 200 bp centered on the mutation; 191 bp upstream of and 10 bp downstream from the mutation; 11 bp upstream of and 190 bp downstream from the mutation. These sequences were retrieved from the human hg18 genome assembly using the UCSC Genome Browser (http://genome.ucsc.edu/) and used as queries in BLAST searches against the masked hg18 genome sequence (BLAST criteria: -F F –b 500 –v 500 –e 1×10−4). The overlap of BLAST hits with functional genes was obtained using the Galaxy online tool (http://main.g2.bx.psu.edu/) (Goecks et al. 2010).
Detection of gene conversion tracts using GENECONV
Acceptor and donor sequences of the 41 high-confidence IGC events were retrieved from the UCSC Genome Browser (http://genome.ucsc.edu/). Disease allele sequences were built by replacing the wild-type sites in the acceptor sequences with the corresponding sites from the donor sequence. The software package GENECONV v.1.81 (Sawyer 1989) was used to identify gene conversion events in multialignments of the acceptor donor and disease allele sequences. DNA stretches of identical nucleotides are recognized as conversion tracts by GENECONV by comparison to the length of identical tracts generated via permutation of the observed alignments. The program assigns a P-value to all possible identical DNA stretches by comparing them to all possible identical stretches in the alignment (global P-value) and each pair of sequences (pairwise P-value). These P-values are corrected for sequence length and also for the number of sequences in the case of global comparison. Of the 41 high-confidence IGC-derived disease alleles, 37 had significant (P-value < 0.05) pairwise tracts and 36 had significant (P-value < 0.05) global tracts that included the pathological mutations. We ran GENECONV using default settings.
Prediction of the deleterious nature of mutations potentially introduced by IGC
The protein sequences of 19,476 human RefSeq genes (3295 HGMD genes and 16,181 non-HGMD genes) were retrieved from the UCSC Genome Browser and used as BLAST queries against the human hg18 masked assembly. All amino acid sites in the query sequence with a different amino acid or stop codon in any subject (paralogous) sequence were collected. We termed these amino acids in the query protein “discordant sites.” Because many paralogous sequences can accumulate frameshifts that are responsible for stretches of low similarity in the BLAST alignments, we retained only discordant sites flanked by at least two nondiscordant amino acids in the three sites immediately upstream and downstream. In addition, we required the 10 amino acids flanking the discordant site either upstream or downstream to share no fewer than six identical sites with the paralogous sequence. Discordant sites that passed our filtering step, and which harbored a stop codon in the corresponding paralogous sequence, were considered to be deleterious; in addition, the potentially deleterious effects of the discordant sites with amino acid replacement were evaluated with PolyPhen-2, which predicts damaging missense mutations using 11 sequence- and structure-based features of proteins and protein alignments (Adzhubei et al. 2010).
Acknowledgments
This work was supported by a grant from the National Science Foundation (DBI-0845494) to M.W.H. and by BIOBASE GmbH (through financial support to D.N.C. and A.D.P.).
References
- ↵IA Adzhubei, S Schmidt, L Peshkin, VE Ramensky, A Gerasimova, P Bork, AS Kondrashov, SR Sunyaev. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249.
- ↵SF Altschul, W Gish, W Miller, EW Myers, DJ Lipman. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410.
- ↵N Arnheim, M Krystal, R Schmickel, G Wilson, O Ryder, E Zimmer. 1980. Molecular evidence for genetic exchanges among ribosomal genes on nonhomologous chromosomes in man and apes. Proc Natl Acad Sci 77: 7323–7327.
- ↵EV Ball, PD Stenson, SS Abeysinghe, M Krawczak, DN Cooper, NA Chuzhanova. 2005. Microdeletions and microinsertions causing human genetic disease: common mechanisms of mutagenesis and the role of local DNA sequence complexity. Hum Mutat 26: 205–213.
- ↵D Benovoy, G Drouin. 2009. Ectopic gene conversions in the human genome. Genomics 93: 27–32.
- ↵JM Bischof, AP Chiang, TE Scheetz, EM Stone, TL Casavant, VC Sheffield, TA Braun. 2006. Genome-wide identification of pseudogenes capable of disease-causing gene conversion. Hum Mutat 27: 545–552.
- ↵P Chen, LJ Xie, GY Huang, XQ Zhao, C Chang. 2005. Mutations of connexin43 in fetuses with congenital heart malformations. Chin Med J (Engl) 118: 971–976.
- ↵JM Chen, DN Cooper, N Chuzhanova, C Férec, GP Patrinos. 2007. Gene conversion: mechanisms, evolution and human disease. Nat Rev Genet 8: 762–775.
- ↵JM Chen, C Ferec, DN Cooper. 2009. Closely spaced multiple mutations as potential signatures of transient hypermutability in human genes. Hum Mutat 30: 1435–1448.
- ↵JM Chen, DN Cooper, C Férec, H Kehrer-Sawatzki, GP Patrinos. 2010. Genomic rearrangements in inherited disease and cancer. Semin Cancer Biol 20: 222–233.
- ↵N Chuzhanova, JM Chen, A Bacolla, GP Patrinos, C Férec, RD Wells, DN Cooper. 2009. Gene conversion causing human inherited disease: evidence for involvement of non-B-DNA-forming sequences and recombination-promoting motifs in DNA breakage and repair. Hum Mutat 30: 1189–1198.
- ↵M Clamp, B Fry, M Kamal, X Xie, J Cuff, MF Lin, M Kellis, K Lindblad-Toh, ES Lander. 2007. Distinguishing protein-coding and noncoding genes in the human genome. Proc Natl Acad Sci 104: 19428–19433.
- ↵DN Cooper, M Krawczak. 1993. Human gene mutation. Bios Scientific Publishers, Oxford, UK.
- ↵DN Cooper, JM Chen, EV Ball, K Howells, M Mort, AD Phillips, N Chuzhanova, M Krawczak, H Kehrer-Sawatzki, PD Stenson. 2010. Genes, mutations, and human inherited disease at the dawn of the age of personalized genomics. Hum Mutat 31: 631–655.
- ↵DN Cooper, A Bacolla, C Ferec, KM Vasquez, H Kehrer-Sawatzki, JM Chen. 2011. On the sequence-directed nature of human gene mutation: The role of genomic architecture and the local DNA sequence environment in mediating gene mutations underlying human inherited disease. Hum Mutat 32: 1075–1099.
- ↵C Dasgupta, AM Martinez, CW Zuppan, MM Shah, LL Bailey, WH Fletcher. 2001. Identification of connexin43 (α1) gap junction gene mutations in patients with hypoplastic left heart syndrome by denaturing gradient gel electrophoresis (DGGE). Mutat Res 479: 173–186.
- ↵K Ezawa, S Oota, N Saitou. 2006. Proceedings of the SMBE Tri-National Young Investigators' Workshop 2005. Genome-wide search of gene conversions in duplicated genes of mouse and rat. Mol Biol Evol 23: 927–940.
- ↵J Goecks, A Nekrutenko, J Taylor. 2010. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11: R86. doi: 10.1186/gb-2010-11-8-r86.
- ↵CH Hsu, Y Zhang, RC Hardison, ED Green, W Miller. 2010. An effective method for detecting gene conversion events in whole genomes. J Comput Biol 17: 1281–1297.
- ↵J Jun, P Ryvkin, E Hemphill, I Mandoiu, C Nelson. 2009. The birth of new genes by RNA- and DNA-mediated duplication during mammalian evolution. J Comput Biol 16: 1429–1444.
- ↵HH Kazazian Jr, C Wong, H Youssoufian, AF Scott, DG Phillips, SE Antonarakis. 1988. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature 332: 164–166.
- ↵AS Kondrashov, IB Rogozin. 2004. Context of deletions and insertions in human coding sequences. Hum Mutat 23: 177–185.
- ↵AS Kondrashov, S Sunyaev, FA Kondrashov. 2002. Dobzhansky-Muller incompatibilities in protein evolution. Proc Natl Acad Sci 99: 14878–14883.
- ↵H Kurahashi, H Inagaki, T Ohye, H Kogo, M Tsutsumi, T Kato, M Tong, BS Emanuel. 2010. The constitutional t(11;22): implications for a novel mechanism responsible for gross chromosomal rearrangements. Clin Genet 78: 299–309.
- ↵JA Lee, CM Carvalho, JR Lupski. 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131: 1235–1247.
- ↵C Lopez-Correa, M Dorschner, H Brems, C Lazaro, M Clementi, M Upadhyaya, D Dooijes, U Moog, H Kehrer-Sawatzki, JL Rutkowski, . 2001. Recombination hotspot in NF1 microdeletion patients. Hum Mol Genet 10: 1387–1392.
- ↵CL McGrath, C Casola, MW Hahn. 2009. Minimal effect of ectopic gene conversion among recent duplicates in four mammalian genomes. Genetics 182: 615–622.
- ↵T Miyata, T Yasunaga, Y Yamawaki-Kataoka, M Obata, T Honjo. 1980. Nucleotide sequence divergence of mouse immunoglobulin γ 1 and γ 2b chain genes and the hypothesis of intervening sequence-mediated domain transfer. Proc Natl Acad Sci 77: 2143–2147.
- ↵PC Ng, S Henikoff. 2003. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31: 3812–3814.
- ↵O Podlaha, J Zhang. 2009. Processed pseudogenes: the ‘fossilized footprints’ of past gene expression. Trends Genet 25: 429–434.
- ↵V Ramensky, P Bork, S Sunyaev. 2002. Human non-synonymous SNPs: server and survey. Nucleic Acids Res 30: 3894–3900.
- ↵S Sawyer. 1989. Statistical tests for detecting gene conversion. Mol Biol Evol 6: 526–538.
- ↵S Scherer, RW Davis. 1980. Recombination of dispersed repeated DNA sequences in yeast. Science 209: 1380–1384.
- ↵DR Schrider, JN Hourmozdi, MW Hahn. 2011. Pervasive multinucleotide mutational events in eukaryotes. Curr Biol 21: 1051–1054.
- ↵PD Stenson, EV Ball, K Howells, AD Phillips, M Mort, DN Cooper. 2009a. The Human Gene Mutation Database: providing a comprehensive central mutation database for molecular diagnostics and personalized genomics. Hum Genomics 4: 69–72.
- ↵PD Stenson, M Mort, EV Ball, K Howells, AD Phillips, NS Thomas, DN Cooper. 2009b. The Human Gene Mutation Database: 2008 update. Genome Med 1: 13. doi: 10.1186/gm13.
- ↵G Terai, A Yoshizawa, H Okida, K Asai, T Mituyama. 2010. Discovery of short pseudogenes derived from messenger RNAs. Nucleic Acids Res 38: 1163–1171.
- ↵D Torrents, M Suyama, E Zdobnov, P Bork. 2003. A genome-wide survey of human pseudogenes. Genome Res 13: 2559–2567.
- ↵AS Waldman, RM Liskay. 1988. Dependence of intrachromosomal recombination in mammalian cells on uninterrupted homology. Mol Cell Biol 8: 5350–5357.
- ↵A Wolf, DS Millar, A Caliebe, M Horan, V Newsway, D Kumpf, K Steinmann, IS Chee, YH Lee, A Mutirangura, . 2009. A gene conversion hotspot in the human growth hormone (GH1) gene promoter. Hum Mutat 30: 239–247.
- ↵Z Zhang, M Gerstein. 2004. Large-scale analysis of pseudogenes in the human genome. Curr Opin Genet Dev 14: 328–335.
Notes
[1] [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.127738.111.