Abstract
Transposable elements (TEs) are a tremendous source of genome instability and genetic variation. Of particular interest to investigators of human biology and human evolution are retrotransposon insertions that are recent and/or polymorphic in the human population. As a consequence, the ability to assay large numbers of polymorphic TEs in a given genome is valuable. Five recent manuscripts each propose methods to scan whole human genomes to identify, map, and, in some cases, genotype polymorphic retrotransposon insertions in multiple human genomes simultaneously. These technologies promise to revolutionize our ability to analyze human genomes for TE-based variation important to studies of human variability and human disease. Furthermore, the approaches hold promise for researchers interested in nonhuman genomic variability. Herein, we explore the methods reported in the manuscripts and discuss their applications to aspects of human biology and the biology of other organisms.
Transposable elements (TEs), comprising two major classes (retrotransposons and DNA transposons), are ubiquitous components of eukaryotic genomes that are often thought of as genomic parasites. They are also powerful agents of evolutionary change. For example, they impact gene expression via the introduction of alternative regulatory elements, exons, and splice junctions (Jurka 1995; Speek 2001; Nigumann et al. 2002; Kazazian 2004; Peaston et al. 2004; Matlik et al. 2006; Babushok et al. 2007; Hasler et al. 2007). However, TEs need not be actively mobilizing to have an effect on genome structure. TE-mediated genome rearrangements through nonhomologous recombination are well-documented (Batzer and Deininger 2002; Lonnig and Saedler 2002; Eichler and Sankoff 2003; Hancks and Kazazian 2010) and deletions, duplications, inversions, translocations, and chromosome breaks have all been linked to the presence of TEs in a variety of genomes (Weil and Wessler 1993; Lim and Simmons 1994; Mathiopoulos et al. 1998; Caceres et al. 1999; Gray 2000; Zhang and Peterson 2004).
The obvious evolutionary question that arises is, “Why are TEs tolerated if they cause so many problems?” Of course, they may simply be too adaptable to be completely eliminated. However, along with recombination, independent assortment, and sex, TE-mediated mutation plays a major role in generating genetic diversity. As potent mutagens, TEs create genetic changes upon which natural selection can act. Their prevalence in eukaryotic genomes may indicate that TEs are, on balance, selectively advantageous and several studies have suggested important roles in genome biology (Vidal et al. 1993; Hamdi et al. 2000; Deininger and Roy-Engel 2002; Nouaud et al. 2003; Lowe et al. 2007; Mikkelsen et al. 2007).
For example, one of the most exciting contributions of TEs to a genome is as a source of raw material in the evolution of new genes and regulatory pathways, aka exaptation or molecular domestication (for examples, see Kapitonov and Jurka 2005; Cordaux et al. 2006b; Feschotte 2008; Lu and Clark 2010; Volff 2010). TEs are recognized as important players in the diversification of taxa by way of their involvement in gene regulation. This point was emphasized with the publication of the Monodelphis domestica (opossum) genome (Mikkelsen et al. 2007) and by numerous other authors (Medstrand et al. 2005; Thornburg et al. 2006; Lowe et al. 2007; Feschotte 2008; Faulkner et al. 2009). For example, in the Monodelphis research the investigators noted that much of the evolutionary innovation distinguishing metatherian from eutherian mammals was not due to differentiation in coding sequences but was instead due to differences in noncoding DNA and that TEs are a “major creative force” in mammalian evolution. Furthermore, one recent publication provided strong arguments suggesting that increases in transposable element activity in response to physiological stress may provide the foundation for the punctuated equilibrium model of evolutionary change (Zeh et al. 2009).
As genetic markers, TEs provide certain advantages over other more widely used systems and have proven to be nearly ideal markers for phylogenetic and population genetic analyses (Murata et al. 1993, 1998; Stoneking et al. 1997; Tatout et al. 1999; Nikaido et al. 2001; Kawai et al. 2002; Xiao et al. 2002; Terai et al. 2003, 2004; Nishihara et al. 2005, 2006; Schmitz et al. 2005; Xing et al. 2005, 2007; Witherspoon et al. 2006; among many others). This is especially true of the retrotransposons, particularly the SINEs (Short INterspersed Elements). First, the presence of an element in multiple individuals at a given locus represents identity by descent in almost all cases because of the very large number of potential insertion sites for any element (Batzer and Deininger 2002; Okada et al. 2004; Ray et al. 2006). Polymorphic TE insertions therefore reflect relationships more accurately than many other genetic markers (e.g., single nucleotide polymorphisms (SNP), microsatellites, and restriction fragment length polymorphisms [RFLP]). In other words, SINEs have been demonstrated to be essentially homoplasy-free (Shedlock et al. 2004; Salem et al. 2005a; Schmitz et al. 2005; Ray et al. 2006). A second advantage is that the ancestral state of a SINE insertion locus is known to be the absence of the element (Perna et al. 1992; Batzer et al. 1994), making assumptions about this aspect of the analysis unnecessary.
Retrotransposons are of particular interest to human biology. They comprise a substantial proportion (∼42%) of the mass of our genome and the only human TE families known to exhibit current mobilization activity (Fig. 1). All three recently active non-LTR retrotransposons in the human genome, LINE-1 (Long INterspersed Element 1, L1), Alu, and SVA have insertions that are human specific and many that are recent enough to still be polymorphic in the human population (Kazazian et al. 1988; Batzer and Deininger 1991, 2002; Batzer et al. 1991; Brouha et al. 2003; Ostertag et al. 2003; Wang et al. 2005). These insertions have tremendous potential to be informative for human biology at a number of levels. Unfortunately, assaying genomes for lineage-specific TE insertions, especially those that are polymorphic among individuals can be a time-consuming and expensive proposition.
Recently active human retrotransposons (Long Terminal Repeat [LTR] and non-LTR groups) and their approximate representation in the human genome (in parentheses). While all sharing a polyA tail, the non-LTR retrotransposons are structurally distinct. The autonomous LINE-1 element (L1) contains two open reading frames while Alu and SVA do not. Alu is instead composed of two monomers linked by an A-rich linker sequence (A5TACA6). SVA is a composite element made up of a hexamer repeat of varying copy number, an Alu-like region, a region of variable numbers of tandem repeats, and an HERV-K derived region known as SINE-R. All non-LTR elements are flanked by target site duplications (arrows) that are typically between 5 and 10 bp. The only recently active LTR element in the human genome (HERV-K) has a distinct structure resembling most endogenous retroviruses—full-length copies contain a central region encoding the Gag, Pol, and Env proteins flanked by identical long terminal repeats and short TSDs. HERV-K was assayed only by Huang et al. (2010), exhibited relatively low insertion rates compared to non-LTR retrotransposons, and will not be mentioned further. L1, Alu, and SVA all mobilize via a mechanism known as TPRT (Target Primed Reverse Transcription; for review, see Ostertag and Kazazian 2001). During this process, the mobilizing element is transcribed via RNA pol II (LINE-1 and SVA) or RNA polIII (Alu). In the case of LINE-1, ORFs 1 and 2 are translated on the ribosomes and ORF1 will typically bind to its own transcript for transport back to the nucleus. Once in the nucleus, ORF1, which has endonuclease and reverse transcriptase activity, is responsible for creating and integrating a cDNA copy at some other location. Alu, and likely SVA elements, “hijack” the L1 enzymatic machinery, probably via docking to the ribosome, in order to facilitate their own nuclear reentry and reverse transcription (Boeke 1997; Ostertag et al. 2003).

Some authors have attempted various experimental methods to identify human-specific TE polymorphisms (Roy et al. 1999; Sheen et al. 2000; Budzin et al. 2002; Badge et al. 2003; Mamedov et al. 2005), but the approaches tended to be rather cumbersome and difficult to optimize. Limited sequencing and computational capacity were also two main problems. As a result, most of the loci used in analyses of human population structure were discovered as part of a disparate set of projects and just happened to be informative with regard to human population differentiation. Thus, their large scale utilization in the scientific community has been rather limited (Bamshad et al. 2003; Watkins et al. 2003).
Fortunately, over the past several years new techniques have revolutionized our ability to generate and analyze DNA sequence data. Whereas only 10 yr ago we were able to generate at best a few hundred thousand bases per day using chain termination sequencing methods, we can now generate gigabases of data in a single run of a 454 (Roche) or Illumina machine. Five recent papers report multiple methods based on second-generation sequencing techniques as well as hybridization arrays to rapidly and relatively inexpensively characterize genome-wide TE insertion patterns (Beck et al. 2010; Ewing and Kazazian 2010b; Huang et al. 2010; Iskow et al. 2010; Witherspoon et al. 2010) and identify a plethora of human TE polymorphisms. With these novel methods, the identification of markers for a vast array of applications can be specifically targeted.
The methods
Many of the methods are novel augmentations of the PCR-based techniques cited above (Roy et al. 1999; Sheen et al. 2000; Budzin et al. 2002; Badge et al. 2003; Mamedov et al. 2005). Ewing and Kazazian (2010b) took advantage of the unique sequence characteristics of the most recently active family of human L1 elements (L1Ta; Kazazian et al. 1988; Skowronski et al. 1988; Kazazian and Moran 1998; Boissinot et al. 2000; Sheen et al. 2000) to generate a library of half-sites (loci containing sequence from an insertion of interest and the neighboring flank) via multiple rounds of PCR. Libraries for 25 individuals including six family groups were then sequenced using Illumina technology to generate a huge data set of ∼12 million 36- or 76-bp single-end reads per individual, that is, ∼20% of a human genome consisting solely of sequences adjacent to recent L1 insertions. These sequence reads were mapped to the human genome reference sequence to identify the locations of the potentially polymorphic L1 insertions.
Similarly, Witherspoon et al. (2010) utilized Illumina technology, but with a different method that targets the genomic sequence junctions of Alu elements. Subsequent steps enriched for Alu-containing PCR amplicons and the resulting libraries were sequenced using a paired-end protocol. Although it could reduce the total number of insertions that could potentially be assayed, using paired-end sequencing gives this method the advantage of having not only a sequence read just upstream of the insertion but also the sequence of the 5′ insertion junction itself, thereby providing a mechanism to verify that the initial round of PCR was due to proper annealing of the Alu-specific primer.
Also taking advantage of the junction between retrotransposons and the adjacent flank were Iskow et al. (2010) in their study of L1 and Alu activity. Using both Sanger sequencing and 454 (Roche) pyrosequencing technology to interrogate the junctions, they investigated insertions in 46 individuals of diverse ancestry to identify 152 novel L1 insertions. Unique to this study, however, was the inclusion of DNA from eight cell lines derived from human tumors, thereby allowing a comparison of activity in normal somatic genomes and genomes thought to be under a differential regulatory regime.
Huang et al. (2010) took a very different approach. Following genome digestion and vectorette PCR, the resulting amplicons were hybridized to a human genome tiling microarray. Analysis of the hybridization data provided information on locations of the sequences flanking L1 insertions in the genomes analyzed.
Finally, Beck et al. (2010) were the only team not to utilize PCR to select for TE insertions in their initial assays. Instead, they used Sanger sequencing to determine the ends of 40 kb fosmid inserts. These end sequences were then used to identify potential size differences in the range of a full-length L1 insertion between these inserts and the human reference sequence. Using this method to survey the genomes of six geographically diverse individuals, they were able to identify 65 insertions not present in the human genome reference or dbRIP, the Database of Retrotransposon Insertion Polymorphisms in Humans (Wang et al. 2006). Furthermore, using cell culture analyses they estimate that each genome contained between three and nine “hot” L1 elements, those with increased activity compared to a previously characterized active LINE, L1.3 (Brouha et al. 2003).
Of course, each approach has its own advantages and disadvantages. For example, by utilizing Illumina sequencing technology, Ewing and Kazazian (2010b) and Witherspoon et al. (2010) were able to scan entire genomes of multiple individuals to identify polymorphisms. However, as is often the case, the cost of so many reads comes in the form of reduced read length and both studies are somewhat limited in their ability to query the human genome reference, especially in highly repetitive regions. Iskow et al. (2010) increased read lengths by utilizing Sanger and pyrosequencing but sacrificed throughput as a consequence. Additionally, for all three of these methods there are also problems with optimization of multiplex sequencing runs and PCR amplification.
The latter optimization problem was overcome by Beck et al. (2010) by eliminating the PCR and instead detecting size differences in large genome fragments. An advantage of this method is the ability to identify full-length insertions at single bp resolution. The major disadvantage, however, is the inability to recognize smaller insertions such as those produced by Alu activity and incomplete reinsertion of L1 elements (most L1 insertions are <1 kb), thereby decreasing throughput. The hybridization (TIP-chip) method of Huang et al. (2010) suffers from both PCR and hybridization optimization problems but this may be offset by the ability to build custom chips for particular genomic regions and the relatively low cost. The individual researcher who considers utilizing any of these methods must choose the appropriate path for his or her laboratory.
Human applications: variation
Many different genetic markers ranging from mitochondrial DNA polymorphisms to microsatellites to SNPs have been applied to investigations of human genetic variation and origins (for reviews, see Relethford 1998; Excoffier 2002; Cavalli-Sforza and Feldman 2003; Pakendorf and Stoneking 2005). Regardless, the ability to assay all or a substantial number of L1, Alu, or SVA insertions in a human genome represents a practical boon to fields related to human genetic variation. One application is to human population genetic and forensic analysis. Because of the homoplasy-free nature of retrotransposon insertions, a number of publications have applied variation in Alu insertion frequencies to ascertaining human demography and its extension, forensic identification of particular individuals or groups. For example, Bamshad et al. (2003), Witherspoon et al. (2006), and Watkins et al. (2003) utilized either Alu or L1 (or a combination of both) to not only explore ancient human origins and migrations but also to cluster continental human populations. Others have extended these results to forensic applications by genotyping unknown individuals and identifying their genetic ancestry with high probability, a potentially useful tool for limiting the field of suspects in a criminal investigation (Ray et al. 2005a).
While these studies have been successful, the identification of novel polymorphisms in the various human populations to provide additional resolution (e.g., intracontinental assignments) has been a difficult task (Mamedov et al. 2005; Cordaux et al. 2007) yielding only a few to a couple of dozen loci per study. However, the studies discussed herein identified numerous insertions with the potential to be useful in this area. For example, Ewing and Kazazian (2010b) identified over 300 nonreference L1 insertions while Witherspoon et al. (2010) simultaneously identified and mapped nearly 500 novel polymorphic Alu insertions in four individuals. Additionally, Beck et al. (2010) identified three L1 insertions apparently restricted to persons of African origin.
Two of the four studies focusing on L1 insertions (Ewing and Kazazian 2010b; Huang et al. 2010) suggest that the current estimates of the rate of L1 insertions in the human genome should be increased. The most recent estimate prior to this work was one new insertion for every 225 births (Xing et al. 2009). Ewing and Kazazian (2010b) and Huang et al. (2010) both essentially doubled this value to between one in 140 births and one in 108 births, respectively. While Beck et al. (2010) did not directly estimate rates of L1 retrotransposition activity, they did note the potential for multiple active L1 elements in all of the genomes surveyed, suggesting the potential for substantial retrotransposition activity. Further support for this idea was provided by Iskow et al. (2010) with their finding that 19% of their population samples exhibited private L1 insertions.
Observing such high rates of L1 mobilization activity is interesting in its own right, but its importance is emphasized when one considers the two other active families of retrotransposons in our genome, Alu and SVA. Both families are considered parasites of L1 and likely rely on L1 for their mobilization (Dewannieux et al. 2003; Ostertag et al. 2003). Alu has been amazingly successful in colonizing our genome (>1 million copies; Lander et al. 2001) and Cordaux et al. (2006a) found an insertion rate for Alu of around one insertion for every 20 births. Of course, these are estimates of overall rates for the human population and do not consider differential rates or the mutational load in individuals, which may vary widely (Brouha et al. 2003; Seleme et al. 2006) or differences in transposition activity between alleles at the same source locus (Lutz et al. 2003). However, in light of the upward revision of our estimates of L1 retrotransposition, should Alu or SVA retrotransposition rates be increased correspondingly? Such a revision is unlikely to be necessary because the estimation methods of Cordaux et al. (2006a) were very different from any of those of any of these studies and therefore independently derived. Unfortunately, Witherspoon et al. (2010) made no attempt to calculate the rate of Alu retrotransposition using their data, likely because they were examining a relatively small subset of Alu elements, the Yb8 and Yb9 subfamilies. No estimates of SVA retrotransposition frequency are available. However, given its likely dependence on L1 enzymatic machinery, the rate of L1 retrotransposition must have some impact on SVA rates.
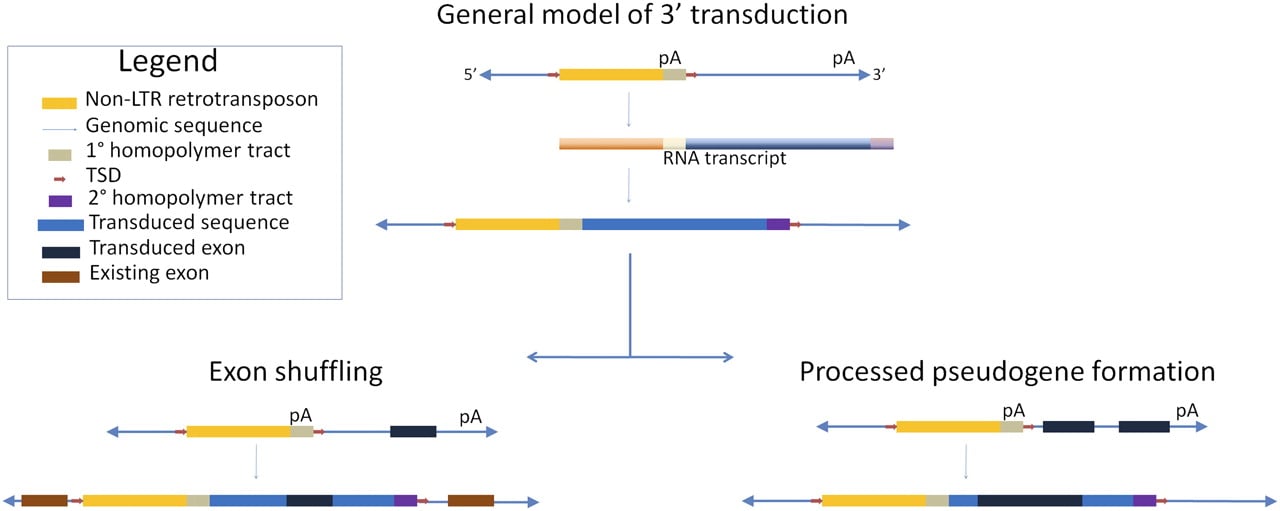
We should not overlook additional human variation impacts of TE-mediated transduction leading to the duplication of portions of the human genome and potentially to exaptation and the formation of novel genes (Fig. 2). Transduction by transposable elements generates genome diversity by exon shuffling (Moran et al. 1999; Goodier et al. 2000; Pickeral et al. 2000; Beck et al. 2010) or through gene family formation (Xing et al. 2006) and at least two of the active human retrotransposon families, LINE-1 and SVA, are known to have participated in transduction events (Holmes et al. 1994; Goodier et al. 2000; Pickeral et al. 2000; Ostertag et al. 2003; Xing et al. 2006). These events provide a means of rapid lineage-specific evolution. The ability to assay all of the polymorphic insertions that may occur between any two individuals allows us the chance to observe evolutionary change in action. Large scale TE display along with powerful computing will allow a direct means to estimate the levels of these types of events within individual genomes and species to determine the contributions that they make to the architecture of the genome. Might some intrepid researchers actually identify a case of exon shuffling or gene duplication due to retrotransposition still segregating in the human population? It is entirely possible given that Beck et al. (2010) noted numerous such transductions ranging from 18 bp to over 1 kb.
Schematic illustrating the mechanism of 3′ transduction by non-LTR retrotransposons and possible gene-related impacts. TE-mediated 3′ transduction occurs when the transcription machinery skips a weak or nonexistent polyadenylation signal (pA). Transcription continues until a downstream polyadenylation signal is recognized. The resulting transcript will contain a portion of the 3′ genomic flank and a secondary homopolymer tract, which will be reverse transcribed into cDNA upon reinsertion into the genome (Boeke and Pickeral 1999; Moran et al. 1999; Goodier et al. 2000). If the transduced sequence contains an exon, it may be inserted near existing exons, resulting in an exon shuffling event. Assuming RNA pol II transcription and normal post-transcriptional processing, two or more exons in the transduced sequence may be merged and reinserted, resulting in a processed pseudogene.
Finally, the recent publication of the pilot paper of the 1000 Genomes Project (http://www.1000genomes.org/; Durbin et al. 2010) and an analysis of the L1 elements in the project data by Ewing and Kazazian (2010a) provides a context for the methods described and the TE variation observed. Briefly, the project's stated aim is to provide deep characterization of human genomic variation and its connection to phenotype. Obviously, any method of sampling comes with some inherent ascertainment bias and the studies described herein are no exception. One of the great strengths of the 1000 Genomes Project is an unbiased comparison of multiple genomes that were all sequenced and assembled in an identical manner. However, initial analysis suggests that the methods discussed by Iskow et al. (2010), Ewing and Kazazian (2010b), and Beck et al. (2010), all of which focused on L1 insertions, have managed to capture snapshots of L1-derived human variation that are very similar to that found by the 1000 Genomes Project. In all cases, nonreference L1 insertions tend to be of relatively low frequency in the human population. Thus, the newly reported methods appear able to accurately ascertain TE diversity in multiple genomes.
Human applications: biomedical
Previous studies have indicated that retrotransposon insertions from all three active families have played a role in the occurrence of human disease either directly, by insertion into or near coding sequences, or indirectly, by serving as loci for nonhomologous recombination (Ostertag and Kazazian 2001; Ostertag et al. 2003; Callinan and Batzer 2006; Cordaux and Batzer 2009). The identification of large numbers of TE insertions with differing levels of variation may provide a new set of markers to deploy in genome-wide association studies (Gibson 2010). Furthermore, the introduction of the new high-throughput ascertainment methods adds a valuable toolkit for identifying potential retrotransposon-based etiologies for de novo instances of genetic disease. For example, in their examination of L1 insertions via the TIP-chip method, Huang et al. (2010) searched specifically for L1 insertions that may be associated with X-linked disorders. While no direct link to a particular pathology was made, at least two insertions with correlations to known human X-linked disorders were indeed observed, suggesting further examination may be needed in these cases.
Somatic retrotransposition events have been identified previously. For example, researchers interested in the mechanism and impact of retrotransposition have engineered L1 elements to demonstrate retrotransposition in somatic cells (Babushok and Kazazian 2007; Garcia-Perez et al. 2007; Coufal et al. 2009; Kano et al. 2009). By including tumor-derived cell lines in their study, Iskow et al. (2010) were able to distinguish germline mutations from those made in somatic cells. Additionally, they were observant enough to note a somatic mobilization in a lung tumor in their small (n = 8) sample of tumor-derived data. Pursuing this outcome, they sampled from additional tumors along with neighboring tissues. Results indicate that lung cancers, in particular, appear to be home to high levels of L1 retrotransposition activity. In all, nine L1 insertions were identified, which when assayed against normal tissues from the same individual, were found to be specific to the tumor. Further analysis suggested that hypomethylation in the tumor cell-lines is at least partially responsible for the increased activity, an observation that is in agreement with numerous studies of L1 regulation (Alves et al. 1996; Jurgens et al. 1996; Yoder et al. 1997; Steinhoff and Schulz 2003; Suter et al. 2004; and Coufal et al. 2009 are several examples from among many). Is this a general pattern for human tumors? Such conclusions are not possible from this study alone due to its limited sample sizes, but other research has suggested that low methylation levels in tumor tissues may allow for increased retrotransposition (for review, see Slotkin and Martienssen 2007).
Extensions to other organisms
While all of the potential discoveries within Homo sapiens represent an exciting prospect, many consider the potential applications to other taxa to be even more exciting. Just as in humans, retrotransposon insertions in other taxa have potential as powerful tools for studying population biology. Most studies of population genetics in nonhuman species are facilitated by mitochondrial DNA, microsatellites, AFLP, or RFLP. Unlike in human studies, SNPs are typically too expensive to use for non-model species and have thus far had limited utility. However, retrotransposon insertions represent a valuable new tool because of their unique combination of genetic properties and the observation that they are one of the least expensive molecular markers to assay. Essentially, all one needs to assay a population is a thermal cycler and gel electrophoresis equipment. Of course, as was the case with humans, developing that all-important library of polymorphic insertions has been a major stumbling block to the widespread use of retrotransposon insertions as population genetic markers (Ray 2007), especially given the paucity of reference genomes from non-model organisms.
However, while each of these studies utilizes the human reference genome to identify specific locations for individual insertions, Witherspoon et al. (2010) point out that with the longer sequence reads now available to users of the Illumina sequencing platform, one could develop a library of polymorphic insertions to “study the population dynamics of nearly any [TE] family in any organism.” As such, this is an opportunity not to be missed by researchers interested in the population dynamics of non-model taxa. It should be noted however, that there may be substantial effort involved in designing and optimizing methods for other taxa. Not the least of these is identifying the polymorphic TE families in a given genome, which can be a daunting prospect. Compiling an inventory of potentially useful retrotransposons is beyond the scope of this commentary. However, for interested researchers, Ohshima and Okada (2005) provided a useful list in their 2005 discussion of LINE/SINE interactions.
Similar applications also exist outside of individual species. For many of the same reasons TEs are good population genetic markers, they also make good markers for the inference of organismal phylogenies (Shedlock and Okada 2000; Okada et al. 2004; Ray et al. 2006). However, the problem of applying the published methods to the identification of insertions polymorphic among taxa could be both more and less difficult. Obviously, there are likely to be multiple polymorphisms when comparing two species that diverged multiple millions of years ago. Thus, finding random differential insertions could be a trivial task. However, because of the evolution of the TEs themselves, a problem could be observed when it comes to identifying informative insertion patterns across the species group.
Researchers familiar with Alu SINEs will be aware that distinct subfamilies of Alu exist in each primate lineage (Carter et al. 2004; Hedges et al. 2004; Otieno et al. 2004; Garber et al. 2005; Ray and Batzer 2005; Ray et al. 2005b; Salem et al. 2005b; Han et al. 2007; Liu et al. 2009; Locke et al. 2011). Each of the methods described relies on sequence characteristics unique to particular subfamilies of elements. Herein lies the problem. When sampling among taxa, should one target particular subfamilies? If so, one may find insertions in one taxon but recover essentially nothing in any other taxa. For example, imagine that a researcher decides to develop a library of polymorphic insertions that will allow them to infer the relationships among humans, chimpanzees, gorillas, and orangutans. He or she unwisely follows Witherspoon's protocol exactly and targets Alu elements from the Yb8/9 family. As a result, they will find a plethora of insertions in the human genome but nothing of interest from any of the other taxa because these families are essentially human specific (Carter et al. 2004; Hedges et al. 2004). The end result will be an unresolved tree because only humans will contain any of the discovered elements.
It is therefore clear that targeting insertions that have been recently active in one taxon may not be the best way to proceed. Instead, one may cast a broader net and target a generalized SINE element from the group of interest. This would likely be a more productive avenue. Again, using humans as a model, we can imagine that the typical primate genome is home to approximately one million Alu insertions. Because of the initial success of Alu early in primate evolution, the majority of these insertions belong to the older subfamilies, J and S (Batzer and Deininger 2002). Thus, when comparing relatively recently diverged taxa, identifying the few hundred or thousand informative insertions will be like searching for the proverbial “needles in the haystack.” Fortunately, modern computational tools may prove to make the problem more tractable and we would encourage interested persons to pursue this as a potential methodology.
Finally, one additional benefit of discovering both population and/or taxon specific insertions is the ability to develop TE-based ascertainment tests for forensic applications to wildlife conservation. A prime example is the investigation into the illegal trade of endangered species. Wildlife conservation often comes into contact only with samples that are not readily identifiable as belonging to one species/population or another. A readily available library of species or population specific markers would be valuable, especially in cases where DNA is limited or degraded (Walker et al. 2003, 2004).
Conclusions
The observations reported in these manuscripts are powerful reminders of the impacts that TEs continue to have on the human genome and have provided valuable information on the way our genomes are being shaped not only in the germline but also in somatic cells, including cells destined to become cancerous. Not only have the investigators given us new perspectives on ongoing retrotransposon activity but they have each developed a new toolkit from which other researchers interested in various aspects of biology, ranging from human disease to endangered species conservation, can select.
Acknowledgments
H. Pagan, J. Smith, V. Joshi, and R. Platt contributed valuable comments to earlier versions of this manuscript. Transposable element research in the Batzer laboratory is supported by the Louisiana Board of Regents Governor's Biotechnology Initiative GBI (2002-005) and National Institutes of Health RO1 GM59290. The Ray laboratory is supported by the Life Sciences Biotechnology Institute and the Mississippi Agricultural and Forestry Experiment Station at Mississippi State University, and by the National Science Foundation (MCB-0841821, DEB-1020865).
References
- ↵G Alves, A Tatro, T Fanning. 1996. Differential methylation of human LINE-1 retrotransposons in malignant cells. Gene 176: 39–44.
- ↵DV Babushok, HH Kazazian Jr. 2007. Progress in understanding the biology of the human mutagen LINE-1. Hum Mutat 28: 527–539.
- ↵DV Babushok, EM Ostertag, HH Kazazian Jr. 2007. Current topics in genome evolution: molecular mechanisms of new gene formation. Cell Mol Life Sci 64: 542–554.
- ↵RM Badge, RS Alisch, JV Moran. 2003. ATLAS: A system to selectively identify human-specific L1 insertions. Am J Hum Genet 72: 823–838.
- ↵MJ Bamshad, S Wooding, WS Watkins, CT Ostler, MA Batzer, LB Jorde. 2003. Human population genetic structure and inference of group membership. Am J Hum Genet 72: 578–589.
- ↵MA Batzer, PL Deininger. 1991. A human-specific subfamily of Alu sequences. Genomics 9: 481–487.
- ↵MA Batzer, PL Deininger. 2002. Alu repeats and human genomic diversity. Nat Rev Genet 3: 370–379.
- ↵MA Batzer, VA Gudi, JC Mena, DW Foltz, RJ Herrera, PL Deininger. 1991. Amplification dynamics of human-specific (HS) Alu family members. Nucleic Acids Res 19: 3619–3623.
- ↵MA Batzer, M Stoneking, M Alegria-Hartman, H Bazan, DH Kass, TH Shaikh, GE Novick, PA Ioannou, WD Scheer, RJ Herrera, . 1994. African origin of human-specific polymorphic Alu insertions. Proc Natl Acad Sci 91: 12288–12292.
- ↵CR Beck, P Collier, C Macfarlane, M Malig, J Kidd, EE Eichler, RM Badge, JV Moran. 2010. LINE-1 retrotransposition activity in human genomes. Cell 141: 1159–1170.
- ↵JD Boeke. 1997. LINEs and Alus–the polyA connection. Nat Genet 16: 6–7.
- ↵JD Boeke, OK Pickeral. 1999. Retroshuffling the genomic deck. Nature 398: 108–109, 111.
- ↵S Boissinot, P Chevret, AV Furano. 2000. L1 (LINE-1) retrotransposon evolution and amplification in recent human history. Mol Biol Evol 17: 915–928.
- ↵B Brouha, J Schustak, RM Badge, S Lutz-Prigge, AH Farley, JV Moran, HH Kazazian Jr. 2003. Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci 100: 5280–5285.
- ↵A Budzin, K Khodosevich, I Mamedov, T Vinogradova, Y Lebedev, G Hunsmann, E Sverdlov. 2002. A technique for genome-wide identification of differences in the interspersed repeats integrations between closely related genomes and its application to detection of human-specific integrations of HERV-K LTRs. Genomics 73: 413–422.
- ↵M Caceres, JM Ranz, A Barbadilla, M Long, A Ruiz. 1999. Generation of a widespread Drosophila inversion by a transposable element. Science 285: 415–418.
- ↵PA Callinan, MA Batzer. 2006. Retrotransposable elements and human disease. Genome Dyn 1: 104–115.
- ↵AB Carter, AH Salem, DJ Hedges, CN Keegan, B Kimball, JA Walker, WS Watkins, LB Jorde, MA Batzer. 2004. Genome-wide analysis of the human Alu Yb-lineage. Hum Genomics 1: 167–178.
- ↵LL Cavalli-Sforza, MW Feldman. 2003. The application of molecular genetic approaches to the study of human evolution. Nat Genet (Suppl) 33: 266–275.
- ↵R Cordaux, MA Batzer. 2009. The impact of retrotransposons on human genome evolution. Nat Rev Genet 10: 691–703.
- ↵R Cordaux, DJ Hedges, SW Herke, MA Batzer. 2006a. Estimating the retrotransposition rate of human Alu elements. Gene 373: 134–137.
- ↵R Cordaux, S Udit, MA Batzer, C Feschotte. 2006b. Birth of a chimeric primate gene by capture of the transposase gene from a mobile element. Proc Natl Acad Sci 103: 8101–8106.
- ↵R Cordaux, D Srikanta, J Lee, M Stoneking, MA Batzer. 2007. In search of polymorphic Alu insertions with restricted geographic distributions. Genomics 90: 154–158.
- ↵NG Coufal, JL Garcia-Perez, GE Peng, GW Yeo, Y Mu, MT Lovci, M Morell, KS O'Shea, JV Moran, FH Gage. 2009. L1 retrotransposition in human neural progenitor cells. Nature 460: 1127–1131.
- ↵PL Deininger, AM Roy-Engel. 2002. Mobile elements in animal and plant genomes. In Mobile DNA II (ed. NL Craig.). ASM Press, Washington, DC.
- ↵M Dewannieux, C Esnault, T Heidmann. 2003. LINE-mediated retrotransposition of marked Alu sequences. Nat Genet 35: 41–48.
- ↵RM Durbin, GR Abecasis, DL Altshuler, A Auton, LD Brooks, RA Gibbs, ME Hurles, GA McVean. 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073.
- ↵EE Eichler, D Sankoff. 2003. Structural dynamics of eukaryotic chromosome evolution. Science 301: 793–797.
- ↵AD Ewing, HH Kazazian. 2010a. Whole-genome resequencing allows detection of many rare LINE-1 insertion alleles in humans. Genome Res doi: 10.1101/gr.114777.110.
- ↵AD Ewing, HH Kazazian Jr. 2010b. High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res 20: 1262–1270.
- ↵L Excoffier. 2002. Human demographic history: refining the recent African origin model. Curr Opin Genet Dev 12: 675–682.
- ↵GJ Faulkner, Y Kimura, CO Daub, S Wani, C Plessy, KM Irvine, K Schroder, N Cloonan, AL Steptoe, T Lassmann, . 2009. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet 41: 563–571.
- ↵C Feschotte. 2008. Transposable elements and the evolution of regulatory networks. Nat Rev Genet 9: 397–405.
- ↵RK Garber, DJ Hedges, SW Herke, NW Hazard, MA Batzer. 2005. The Alu Yc1 subfamily: Sorting the wheat from the chaff. Cytogenet Genome Res 110: 537–542.
- ↵JL Garcia-Perez, MC Marchetto, AR Muotri, NG Coufal, FH Gage, KS O'Shea, JV Moran. 2007. LINE-1 retrotransposition in human embryonic stem cells. Hum Mol Genet 16: 1569–1577.
- ↵G Gibson. 2010. Hints of hidden heritability in GWAS. Nat Genet 42: 558–560.
- ↵JL Goodier, EM Ostertag, HH Kazazian Jr. 2000. Transduction of 3′-flanking sequences is common in L1 retrotransposition. Hum Mol Genet 9: 653–657.
- ↵YH Gray. 2000. It takes two transposons to tango: transposable-element-mediated chromosomal rearrangements. Trends Genet 16: 461–468.
- ↵HK Hamdi, H Nishio, J Tavis, R Zielinski, A Dugaiczyk. 2000. Alu-mediated phylogenetic novelties in gene regulation and development. J Mol Biol 299: 931–939.
- ↵K Han, MK Konkel, J Xing, H Wang, J Lee, TJ Meyer, CT Huang, E Sandifer, K Hebert, EW Barnes, . 2007. Mobile DNA in Old World monkeys: a glimpse through the rhesus macaque genome. Science 316: 238–240.
- ↵DC Hancks, HH Kazazian Jr. 2010. SVA retrotransposons: evolution and genetic instability. Semin Cancer Biol 20: 234–245.
- ↵J Hasler, T Samuelsson, K Strub. 2007. Useful “junk”: Alu RNAs in the human transcriptome. Cell Mol Life Sci 64: 1793–1800.
- ↵DJ Hedges, PA Callinan, R Cordaux, J Xing, E Barnes, MA Batzer. 2004. Differential Alu mobilization and polymorphism among the human and chimpanzee lineages. Genome Res 14: 1068–1075.
- ↵SE Holmes, BA Dombroski, CM Krebs, CD Boehm, HH Kazazian Jr. 1994. A new retrotransposable human L1 element from the LRE2 locus on chromosome 1q produces a chimaeric insertion. Nat Genet 7: 143–148.
- ↵CRL Huang, AM Schneider, Y Lu, T Nirangan, P Shen, MA Robinson, JP Steranka, D Valle, CI Civin, T Wang, . 2010. Mobile interspersed repeats are major structural variants in the human genome. Cell 141: 1171–1182.
- ↵RC Iskow, MT McCabe, RE Mills, S Torene, WS Pittard, AF Neuwald, EG Van Meir, PM Vertino, SE Devine. 2010. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell 141: 1253–1261.
- ↵B Jurgens, BJ Schmitz-Drager, WA Schulz. 1996. Hypomethylation of L1 LINE sequences prevailing in human urothelial carcinoma. Cancer Res 56: 5698–5703.
- ↵J Jurka. 1995. Origin and evolution of Alu repetitive elements. In Impact of short interspersed elements (SINEs) on the host genome (ed. RJ Maraia), pp. 25–41. Landes Company, Austin, TX.
- ↵H Kano, I Godoy, C Courtney, MR Vetter, GL Gerton, EM Ostertag, HH Kazazian Jr. 2009. L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes Dev 23: 1303–1312.
- ↵VV Kapitonov, J Jurka. 2005. RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol 3: e181. doi: 10.1371/journal.pbio.0030181.
- ↵K Kawai, M Nikaido, M Harada, S Matsumura, LK Lin, Y Wu, M Hasegawa, N Okada. 2002. Intra- and interfamily relationships of Vespertilionidae inferred by various molecular markers including SINE insertion data. J Mol Evol 55: 284–301.
- ↵HH Kazazian Jr. 2004. Mobile elements: drivers of genome evolution. Science 303: 1626–1632.
- ↵HH Kazazian Jr, JV Moran. 1998. The impact of L1 retrotransposons on the human genome. Nat Genet 19: 19–24.
- ↵HH Kazazian Jr, C Wong, H Youssoufian, AF Scott, DG Phillips, SE Antonarakis. 1988. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature 332: 164–166.
- ↵ES Lander, LM Linton, B Birren, C Nusbaum, MC Zody, J Baldwin, K Devon, K Dewar, M Doyle, W FitzHugh, . 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921.
- ↵JK Lim, MJ Simmons. 1994. Gross chromosome rearrangements mediated by transposable elements in Drosophila melanogaster. Bioessays 16: 269–275.
- ↵GE Liu, C Alkan, L Jiang, S Zhao, EE Eichler. 2009. Comparative analysis of Alu repeats in primate genomes. Genome Res 19: 876–885.
- ↵DP Locke, LW Hillier, WC Warren, KC Worley, LV Nazareth, DM Muzny, S-P Yang, Z Wang, AT Chinwalla, P Minx, . 2011. Comparative and demographic analysis of orangutan genomes. Nature 469: 529–533.
- ↵WE Lonnig, H Saedler. 2002. Chromosome rearrangements and transposable elements. Annu Rev Genet 36: 389–410.
- ↵CB Lowe, G Bejerano, D Haussler. 2007. Thousands of human mobile element fragments undergo strong purifying selection near developmental genes. Proc Natl Acad Sci 104: 8005–8010.
- ↵J Lu, AG Clark. 2010. Population dynamics of PIWI-interacting RNAs (piRNAs) and their targets in Drosophila. Genome Res 20: 212–227.
- ↵SM Lutz, BJ Vincent, HH Kazazian Jr, MA Batzer, JV Moran. 2003. Allelic heterogeneity in LINE-1 retrotransposition activity. Am J Hum Genet 73: 1431–1437.
- ↵IZ Mamedov, ES Arzumanyan, AL Amosova, YB Lebedev, ED Sverdlov. 2005. Whole-genome experimental identification of insertion/deletion polymorphisms of interspersed repeats by a new general approach. Nucleic Acids Res 33: e16. doi: 10.1093/nar/gni018.
- ↵KD Mathiopoulos, A della Torre, V Predazzi, V Petrarca, M Coluzzi. 1998. Cloning of inversion breakpoints in the Anopheles gambiae complex traces a transposable element at the inversion junction. Proc Natl Acad Sci 95: 12444–12449.
- ↵K Matlik, K Redik, M Speek. 2006. L1 antisense promoter drives tissue-specific transcription of human genes. J Biomed Biotechnol 1: 1–16.
- ↵P Medstrand, LN van de Lagemaat, CA Dunn, JR Landry, D Svenback, DL Mager. 2005. Impact of transposable elements on the evolution of mammalian gene regulation. Cytogenet Genome Res 110: 342–352.
- ↵TS Mikkelsen, MJ Wakefield, B Aken, CT Amemiya, JL Chang, S Duke, M Garber, AJ Gentles, L Goodstadt, A Heger, . 2007. Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature 447: 167–177.
- ↵JV Moran, RJ DeBerardinis, HH Kazazian Jr. 1999. Exon shuffling by L1 retrotransposition. Science 283: 1530–1534.
- ↵S Murata, N Takasaki, M Saitoh, N Okada. 1993. Determination of the phylogenetic relationships among Pacific salmonids by using short interspersed elements (SINEs) as temporal landmarks of evolution. Proc Natl Acad Sci 90: 6995–6999.
- ↵S Murata, N Takasaki, T Okazaki, T Kobayashi, K Numachi, K-H Chang, N Okada. 1998. Molecular evidence from short interspersed elements (SINEs) that Onchorhynchus masou (cherry salmon) is monophyletic. Can J Fish Aquat Sci 55: 1864–1870.
- ↵P Nigumann, K Redik, K Matlik, M Speek. 2002. Many human genes are transcribed from the antisense promoter of L1 retrotransposon. Genomics 79: 628–634.
- ↵M Nikaido, F Matsuno, H Hamilton, RL Brownell Jr, Y Cao, W Ding, Z Zuoyan, AM Shedlock, RE Fordyce, M Hasegawa, . 2001. Retroposon analysis of major cetacean lineages: The monophyly of toothed whales and the paraphyly of river dolphins. Proc Natl Acad Sci 98: 7384–7389.
- ↵H Nishihara, Y Satta, M Nikaido, JGM Thewissen, MJ Stanhope, N Okada. 2005. A retroposon analysis of Afrotherian phylogeny. Mol Biol Evol 22: 1823–1833.
- ↵H Nishihara, M Hasegawa, N Okada. 2006. Pegasoferae, an unexpected mammalian clade revealed by tracking ancient retroposon insertions. Proc Natl Acad Sci 103: 9929–9934.
- ↵D Nouaud, H Quesneville, D Anxolabehere. 2003. Recurrent exon shuffling between distant P-element families. Mol Biol Evol 20: 190–199.
- ↵K Ohshima, N Okada. 2005. SINEs and LINEs: symbionts of eukaryotic genomes with a common tail. Cytogenet Genome Res 110: 475–490.
- ↵N Okada, AM Shedlock, M Nikaido. 2004. Retroposon mapping in molecular systematics. In Mobile genetic elements: Protocols and genomic applications (ed. WJ Miller, P Capy), Vol. 260, pp. 189–226. Humana Press, Totowa, NJ.
- ↵EM Ostertag, HH Kazazian Jr. 2001. Biology of mammalian L1 retrotransposons. Annu Rev Genet 35: 501–538.
- ↵EM Ostertag, JL Goodier, Y Zhang, HH Kazazian Jr. 2003. SVA elements are nonautonomous retrotransposons that cause disease in humans. Am J Hum Genet 73: 1444–1451.
- ↵AC Otieno, AB Carter, DJ Hedges, JA Walker, DA Ray, RK Garber, BA Anders, N Stoilova, ME Laborde, JD Fowlkes, . 2004. Analysis of the human Alu Ya-lineage. J Mol Biol 342: 109–118.
- ↵B Pakendorf, M Stoneking. 2005. Mitochondrial DNA and human evolution. Annu Rev Genomics Hum Genet 6: 165–183.
- ↵AE Peaston, AV Evsikov, JH Graber, WN de Vries, AE Holbrook, D Solter, BB Knowles. 2004. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev Cell 7: 597–606.
- ↵NT Perna, MA Batzer, PL Deininger, M Stoneking. 1992. Alu insertion polymorphism: a new type of marker for human population studies. Hum Biol 64: 641–648.
- ↵OK Pickeral, W Makalowski, MS Boguski, JD Boeke. 2000. Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Res 10: 411–415.
- ↵DA Ray. 2007. SINEs of progress: Mobile element applications to molecular ecology. Mol Ecol 16: 19–33.
- ↵DA Ray, MA Batzer. 2005. Tracking Alu evolution in New World primates. BMC Evol Biol 5: 51. doi: 10.1186/1471-2148-5-51.
- ↵DA Ray, JA Walker, A Hall, B Llewellyn, J Ballantyne, AT Christian, K Turteltaub, MA Batzer. 2005a. Inference of human geographic origins using Alu insertion polymorphisms. Forensic Sci Int 153: 117–124.
- ↵DA Ray, J Xing, DJ Hedges, MA Hall, ME Laborde, BA Anders, BR White, N Stoilova, JD Fowlkes, KE Landry, . 2005b. Alu insertion loci and platyrrhine primate phylogeny. Mol Phylogenet Evol 35: 117–126.
- ↵DA Ray, J Xing, A-H Salem, MA Batzer. 2006. SINEs of a nearly perfect character. Syst Biol 55: 928–935.
- ↵JH Relethford. 1998. Genetics of modern human origins and diversity. Annu Rev Anthropol 27: 1–23.
- ↵AM Roy, ML Carroll, DH Kass, SV Nguyen, AH Salem, MA Batzer, PL Deininger. 1999. Recently integrated human Alu repeats: finding needles in the haystack. Genetica 107: 149–161.
- ↵AH Salem, DA Ray, MA Batzer. 2005a. Identity by descent and DNA sequence variation of human SINE and LINE elements. Cytogenet Genome Res 108: 63–72.
- ↵AH Salem, DA Ray, DJ Hedges, J Jurka, MA Batzer. 2005b. Analysis of the human Alu Ye lineage. BMC Evol Biol 5: 18. doi: 10.1186/1471-2148-5.
- ↵J Schmitz, C Roos, H Zischler. 2005. Primate phylogeny: molecular evidence from retroposons. Cytogenet Genome Res 108: 26–37.
- ↵MC Seleme, MR Vetter, R Cordaux, L Bastone, MA Batzer, HH Kazazian Jr. 2006. Extensive individual variation in L1 retrotransposition capability contributes to human genetic diversity. Proc Natl Acad Sci 103: 6611–6616.
- ↵AM Shedlock, N Okada. 2000. SINE insertions: powerful tools for molecular systematics. Bioessays 22: 148–160.
- ↵AM Shedlock, K Takahashi, N Okada. 2004. SINEs of speciation: tracking lineages with retroposons. Trends Ecol Evol 19: 545–553.
- ↵FM Sheen, ST Sherry, GM Risch, M Robichaux, I Nasidze, M Stoneking, MA Batzer, GD Swergold. 2000. Reading between the LINEs: human genomic variation induced by LINE-1 retrotransposition. Genome Res 10: 1496–1508.
- ↵J Skowronski, TG Fanning, MF Singer. 1988. Unit-length line-1 transcripts in human teratocarcinoma cells. Mol Cell Biol 8: 1385–1397.
- ↵RK Slotkin, R Martienssen. 2007. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 8: 272–285.
- ↵M Speek. 2001. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Mol Cell Biol 21: 1973–1985.
- ↵C Steinhoff, WA Schulz. 2003. Transcriptional regulation of the human LINE-1 retrotransposon L1.2B. Mol Genet Genomics 270: 394–402.
- ↵M Stoneking, JJ Fontius, SL Clifford, H Soodyall, SS Arcot, N Saha, T Jenkins, MA Tahir, PL Deininger, MA Batzer. 1997. Alu insertion polymorphisms and human evolution: evidence for a larger population size in Africa. Genome Res 7: 1061–1071.
- ↵CM Suter, DI Martin, RL Ward. 2004. Hypomethylation of L1 retrotransposons in colorectal cancer and adjacent normal tissue. Int J Colorectal Dis 19: 95–101.
- ↵C Tatout, S Warwick, A Lenoir, JM Deragon. 1999. SINE insertions as clade markers for wild crucifer species. Mol Biol Evol 16: 1614–1621.
- ↵Y Terai, K Takahashi, M Nishida, T Sato, N Okada. 2003. Using SINEs to probe ancient explosive speciation: “Hidden” radiation of African cichlids? Mol Biol Evol 20: 924–930.
- ↵Y Terai, N Takezaki, WE Mayer, H Tichy, N Takahata, J Klein, N Okada. 2004. Phylogenetic relationships among East African haplochromine fish as revealed by short interspersed elements (SINEs). J Mol Evol 58: 64–78.
- ↵BG Thornburg, V Gotea, W Makalowski. 2006. Transposable elements as a significant source of transcription regulating signals. Gene 365: 104–110.
- ↵F Vidal, E Mougneau, N Glaichenhaus, P Vaigot, M Darmon, F Cuzin. 1993. Coordinated posttranscriptional control of gene expression by modular elements including Alu-like repetitive sequences. Proc Natl Acad Sci 90: 208–212.
- ↵JN Volff. 2010. Tame affairs: domesticated transposase and domestic pigs. EMBO Rep 11: 241–242.
- ↵JA Walker, DA Hughes, BA Anders, J Shewale, SK Sinha, MA Batzer. 2003. Quantitative intra-short interspersed element PCR for species-specific DNA identification. Anal Biochem 316: 259–269.
- ↵JA Walker, DA Hughes, DJ Hedges, BA Anders, ME Laborde, J Shewale, SK Sinha, MA Batzer. 2004. Quantitative PCR for DNA identification based on genome-specific interspersed repetitive elements. Genomics 83: 518–527.
- ↵H Wang, J Xing, D Grover, DJ Hedges, K Han, JA Walker, MA Batzer. 2005. SVA elements: a hominid-specific retroposon family. J Mol Biol 354: 994–1007.
- ↵J Wang, L Song, D Grover, S Azrak, MA Batzer, P Liang. 2006. dbRIP: A highly integrated database of retrotransposon insertion polymorphisms in humans. Hum Mutat 27: 323–329.
- ↵WS Watkins, AR Rogers, CT Ostler, S Wooding, MJ Bamshad, AM Brassington, ML Carroll, SV Nguyen, JA Walker, BV Prasad, . 2003. Genetic variation among world populations: inferences from 100 Alu insertion polymorphisms. Genome Res 13: 1607–1618.
- ↵CF Weil, SR Wessler. 1993. Molecular evidence that chromosome breakage by Ds elements is caused by aberrant transposition. Plant Cell 5: 515–522.
- ↵DJ Witherspoon, EE Marchani, WS Watkins, CT Ostler, SP Wooding, BA Anders, JD Fowlkes, S Boissinot, AV Furano, DA Ray, . 2006. Human population genetic structure and diversity inferred from polymorphic L1(LINE-1) and Alu insertions. Hum Hered 62: 30–46.
- ↵DJ Witherspoon, J Xing, Y Zhang, WS Watkins, MA Batzer, LB Jorde. 2010. Mobile element scanning (ME-Scan) by targeted high-throughput sequencing. BMC Genomics 11: 410. doi: 10.1186/1471-2164-11-410.
- ↵FX Xiao, JF Yang, JJ Cassiman, R Decorte. 2002. Diversity at eight polymorphic Alu insertion loci in Chinese populations shows evidence for European admixture in an ethnic minority population from northwest China. Hum Biol 74: 555–568.
- ↵J Xing, H Wang, K Han, DA Ray, CH Huang, LG Chemnick, CB Stewart, TR Disotell, OA Ryder, MA Batzer. 2005. A mobile element based phylogeny of Old World monkeys. Mol Phylogenet Evol 37: 872–880.
- ↵J Xing, H Wang, VP Belancio, R Cordaux, PL Deininger, MA Batzer. 2006. Emergence of primate genes by retrotransposon-mediated sequence transduction. Proc Natl Acad Sci 103: 17608–17613.
- ↵J Xing, H Wang, Y Zhang, DA Ray, AJ Tosi, TR Disotell, MA Batzer. 2007. A mobile element-based evolutionary history of guenons (tribe Cercopithecini). BMC Biol 5: 5.
- ↵J Xing, Y Zhang, K Han, AH Salem, SK Sen, CD Huff, Q Zhou, EF Kirkness, S Levy, MA Batzer, . 2009. Mobile elements create structural variation: Analysis of a complete human genome. Genome Res 19: 1516–1526.
- ↵JA Yoder, CP Walsh, TH Bestor. 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet 13: 335–340.
- ↵DW Zeh, JA Zeh, Y Ishida. 2009. Transposable elements and an epigenetic basis for punctuated equilibria. Bioessays 31: 715–726.
- ↵J Zhang, T Peterson. 2004. Transposition of reversed Ac element ends generates chromosome rearrangements in maize. Genetics 167: 1929–1937.
Notes
[1] Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.110528.110.