Abstract
Fields such as genomics and systems biology are built on the synergism between computational and experimental techniques. This type of synergism is especially important in accomplishing goals like identifying all functional transcription factor binding sites in vertebrate genomes. Precise detection of these elements is a prerequisite to deciphering the complex regulatory networks that direct tissue specific and lineage specific patterns of gene expression. This review summarizes approaches for in silico, in vitro, and in vivo identification of transcription factor binding sites. A variety of techniques useful for localized- and high-throughput analyses are discussed here, with emphasis on aspects of data generation and verification.
One documented goal of the National Human Genome Research Institute (NHGRI) is the identification of all functional noncoding elements in the human genome (ENCODE Project Consortium 2004). Studies by ENCODE Consortium members and other investigators in the field have demonstrated that a mixture of computational and experimental approaches is required for the genome-wide elucidation of cis-acting transcriptional regulatory elements. These include promoters, enhancers, and repressor elements, along with structural components like origins of replication and boundary elements. For instance, experimentally based oligo-capping methods represent technical advances toward defining the precise 5′ ends of mRNA transcripts (Suzuki et al. 2002; Shiraki et al. 2003; for review, see Harbers and Carninci 2005), enabling the robust prediction of proximal promoter regions and their components (Trinklein et al. 2003; Cooper et al. 2006). In addition, sensitive and comprehensive microarray-based analyses of human RNAs are providing a detailed map of the transcribed regions of the human genome (ENCODE Consortium, in prep.). Methods to characterize replication origins on a genome-wide scale are also in development. Techniques like microarrays are providing details on the coordinated timing of replication by detecting twofold increases in DNA copy number, or heavy isotope incorporation into newly synthesized DNA (for review, see Schwob 2004; MacAlpine and Bell 2005; ENCODE Consortium, in prep.). Along with newly emerging techniques, a few historically proven approaches still provide reliable indicators of functional regions. These include the detection of altered chromatin structure using DNaseI hypersensitivity (Weisbrod and Weintraub 1979; ENCODE Consortium, in prep.) and sequence conservation as found in pairwise- or multi-species comparisons (for review, see Miller et al. 2004; ENCODE Consortium, in prep.).
The function of promoters, enhancers, replication origins, and other regulatory elements is mediated by DNA/protein interactions. Thus, one major step in the characterization of the functional elements of the human genome is the identification of all the protein binding sites, which serve as the atomic units of functional activity (Collins 2003). Recent studies focused on the analysis of transcription factor binding sites in one percent of the human genome (ENCODE Consortium, in prep.) have revealed the need for integrated computational and experimental approaches in the identification of genome-scale sets of transcriptional regulatory elements. Given the amount of noncoding sequence that is under selective constraint (∼3.5% of the human genome; Waterston et al. 2002; Chiaromonte et al. 2003), the anticipated number of DNA binding factors (∼1962; Messina et al. 2004), the complexity of finding a suitable biological assay to detect a given functional activity, and the cost of pursuing such efforts, synergistic collaborations between computational prediction and high-throughput experimental validation remain a critical necessity.
The techniques summarized herein are meant to provide a comprehensive overview of the complementary aspects of computational prediction and experimental validation of functional sites. These described methods are often considered fundamental to investigators working within that specific field but may be unfamiliar to an outside investigator, such as a biologist wanting to predict the identity of transcription factor binding sites (TFBSs), or a computer programmer trying to validate the prediction of a biological feature. In that spirit, we include techniques from categories that address locus-specific and high-throughput methodologies. Supplemental tables list the Web servers available for computational predictions and provide URLs for protocols of experimental techniques. Additionally, readers are encouraged to examine the journal Nucleic Acids Research for its Annual Review of Bioinformatics Web Sites (2006, http://nar.oxfordjournals.org/).
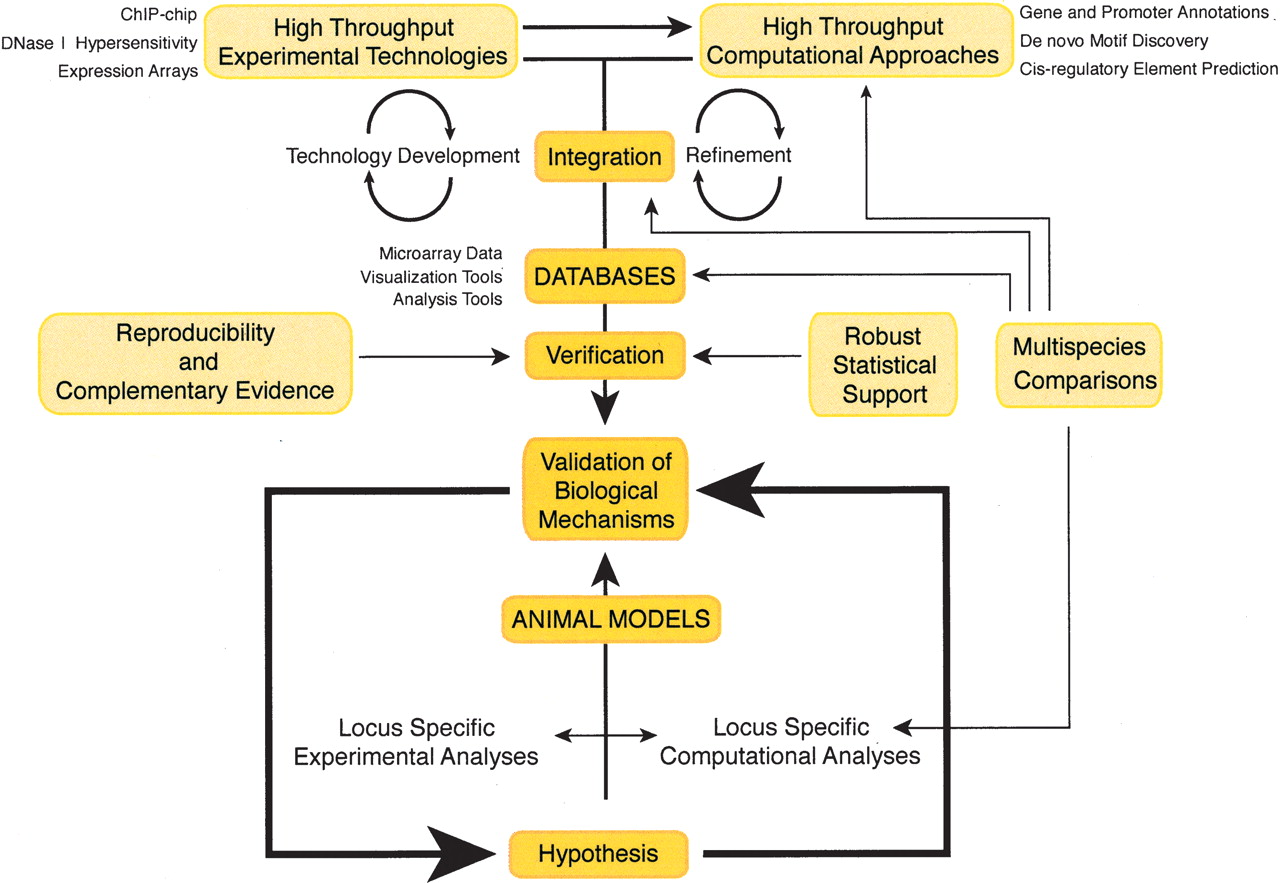
Although broad in scope, this review of computational and experimental techniques is intended to elucidate aspects of their interdependence. Computational techniques, by definition, are predictive and vary in performance quality. Experimental results provide a spectrum of information, ranging from implied functional relevance to validation of protein identity. In this review, we will begin with a description of computational approaches used to identify transcription factor binding sites, define a need for additional experimental data sets, and introduce several experimental methodologies for identifying regulatory elements. We will then end with a description of how computational analyses of these experimental data sets can provide new insights into transcriptional regulation. Figure 1 illustrates this interplay between computational and experimental strategies. Whether initiating from a locus-specific or high-throughput perspective, all indicated pathways lead to the ultimate goal of validation of a biological mechanism.
Computational techniques
A computational approach to studying transcriptional regulatory networks requires the analysis of large and complex data sets. These data sets often include such diverse yet interdependent data as (1) gene expression profiles, (2) locations of promoters and computationally predicted transcription factor binding sites, (3) experimentally identified target genes of specific transcription factor families, and (4) sequence conservation (for review, see Qiu 2003). Using such data sets, investigators have produced, for example, a computational catalog of high-quality putative regulatory elements from vertebrates (Prakash and Tompa 2005). Also, ab initio approaches using the techniques of conservation, overrepresentation, and coregulation are successfully being applied to determine cohorts of expression groups within the genome (Cora et al. 2005). Methods to identify tissue-specific factors have evolved from detecting single factors that regulate expression in tissues such as liver (Krivan and Wasserman 2001) or muscle (Wasserman and Fickett 1998) to comprehensively identifying novel motifs that confer tissue-specific expression patterns (Qian et al. 2005; Blanchette et al. 2006; Huber and Bulyk 2006). As transcription factors often work cooperatively, binding in close physical proximity, recent computational approaches have used the presence of co-occurring motifs to identify putative regulatory modules (Kreiman 2004; Zhou and Wong 2004; Zhu et al. 2005). The recent analysis by Blanchette and colleagues (2006) predicted more than 118,000 such regulatory modules in the human genome. Clearly, these (and other) computational approaches used to identify transcription networks are providing new insights into transcriptional regulation. However, in this review, we will focus only on the various computational methodologies used to predict transcription factor binding sites. An in-depth discussion of all the computational techniques used to predict binding sites is beyond the scope of this review; however, a survey of tools available as Web-based resources is documented in Supplemental Table 1. Also, evaluations of some computational tools are available in other publications (Roulet et al. 1998; Tompa et al. 2005).
The myriad approaches to the computational prediction of functional binding sites are all based on either pattern matching or pattern detection (Supplemental Table 1). Pattern matching utilizes prior knowledge of all characterized DNA binding sites for a given protein. Finding these patterns within the genome allows one to identify putative protein binding sites that might represent uncharacterized regulatory elements (van Helden 2003). Pattern matching requires that the known binding sites for a given protein be represented as a consensus of the collection or as a matrix of acceptable nucleotides at each position. The use of consensus patterns is convenient but may exclude a significant subset of a binding site repertoire because of omission of important variable regions (Roulet et al. 1998). Some of the missing information can be captured through IUPAC strings (an alphabet of characters other than ACGT) that provide a means to indicate alternative choices at each position (e.g., Y = C or T). However, IUPAC strings, while conveying more information than a core consensus, do not provide information as to the relative frequency of the alternative nucleotides. The information content of regulatory sites can be more accurately represented by position weight matrices (PWMs) (Stormo et al. 1982; Harr et al. 1983) or position-specific scoring matrices (PSSMs), which incorporate pattern variability by recording the frequencies of nucleotides at each site or by assigning penalties to nucleotides that should not appear within a factor binding site. Although PWM pattern matching represents an improvement over consensus mapping in sensitivity (i.e., it has a lower false negative rate), when used as the sole means of identifying protein-binding sites it still suffers from the limited amount of training data available (Roulet et al. 1998) and often results in a high rate of false-positive predictions (Tompa et al. 2005; Jolly et al. 2005).
A number of algorithmic approaches have been developed for de novo pattern detection (i.e., the discovery of unknown motifs), many of which search for recurring or overrepresented patterns in DNA. Examples include Hidden Markov Models (Pedersen and Moult 1996), Gibbs sampling (Lawrence et al. 1993), greedy alignment algorithms (e.g., CONSENSUS, Hertz and Stormo 1999), expectation-maximization (MEME, Bailey and Elkan 1995), probabilistic mixture modeling (NestedMica, Down and Hubbard 2005) and exhaustive enumeration (i.e., detecting the set of all nucleotide n-mers, then reporting the most frequent or overrepresented; e.g., Weeder, Pavesi et al. 2004). Alternatively, variations of a pattern can be modeled using information theory (Schneider 2000). Using this approach, the frequencies of nucleotides at each position give insight into whether a protein binds to the major or minor groove of the DNA helix. Once these patterns are determined for a particular protein, the range of variation in target binding sequences can be modeled and matched to the genome (Gadiraju et al. 2003; Vyhlidal et al. 2004).
The use of orthologous sequences, also referred to as phylogenetic footprinting, introduces the filtering power of evolutionary constraint to identify putative regulatory regions that stand apart from the background sequence conservation (Tagle et al. 1988). The search for both known binding sites (pattern matching) and overrepresented novel motifs (pattern detection) can be improved through the analysis of data sets containing orthologous or coregulated genes (summarized in Frith et al. 2004). In one case, regions that colocalized as high-scoring PWM matches and conserved regions in human–mouse–rat genomic alignments provided a 44-fold increase in the specificity of the predictions compared with pattern matching alone (Gibbs et al. 2004). In a study involving pattern detection, Xie et al. (2005) report the first comprehensive screen for regulatory motifs in human promoters by identifying motifs that are enriched above background and are conserved in human, mouse, rat, and dog genomes. Analysis tools that have been refined by incorporating cross-species conservation include Gibbs sampling (e.g., CompareProspector, Liu et al. 2004; PhyloGibbs, Siddharthan et al. 2004, 2005); expectation maximization (PhyloME, Sinha et al. 2003; orthoMEME, Sinha et al. 2003; EMnEM, Moses et al. 2004) and greedy alignment algorithms (PhyloCon, Wang and Stormo 2003). A variation of phylogenetic footprinting known as phylogenetic shadowing uses the collective divergence time of a relatively large number of closely related species (Boffelli et al. 2003). This has the advantage of identifying functional elements that are specific to a lineage from within an unambiguously aligned set of sequences. The disadvantage of this approach lies in the fact that the number of genomic sequences required for such an analysis is currently prohibitive for most investigators.
The recent refinements of computational techniques for identifying binding sites have evoked considerable interest from the field in the development of follow-up or validation analyses. For example, evolutionary constraint has been used not only to identify sites but also to distinguish real motifs from false positives (Blanchette and Sinha 2001) and to discern potentially functional sites from neutral DNA (King et al. 2005). Other validation analyses capitalize on properties of regulatory elements such as the presence of spaced dyads (pairs of short words separated by a fixed distance) and the propensity for palindromic content (van Helden et al. 2000), as well as the interdependence of bases at specific positions within a motif (Wang et al. 2005). Also, the information content and binding preferences of known motifs have been used to identify binding sites of new family members (Keles et al. 2003). Currently, the limiting factor in confirmation and refinement of in silico predictions is a lack of experimental data (Vavouri and Elgar 2005). Despite our best efforts at predicting functional sites, the cellular environment dictates which events can and cannot occur by imposing the selective constraint of higher-order chromatin structure; consequently, experimental confirmation remains the highest form of validation. Described below are various experimental techniques that can be used in conjunction with the computational approaches depicted above.
Experimental techniques
Experimental approaches to identifying transcription factor binding sites are necessary to understand their contributions to biological function, to address the complexity of tissue-specific and temporal stage-specific effects on gene expression (Levine and Tjian 2003), and to continue refinement of computational predictions. Experimental techniques useful for identifying transcription factor binding sites include those that, although not directly measuring transcription factor/DNA interactions, can lead to the identification of regulatory elements. Such techniques include analysis of alterations of chromatin structure and experimental manipulation of defined DNA segments, both of which are advantageous in helping to locate a functional element when the exact regulatory protein(s) involved is not known. Other techniques, which directly measure protein/DNA interactions, provide more precise information but are only useful after the identity of the critical transcription factor has been established. Examples of these two types of approaches, each of which can range in scope from localized, site-specific analyses to high-throughput assays that generate broad conclusions about binding site preferences and regulation of gene expression, are described below. Protocols for these experimental assays are available in the references and in Supplemental Table 2.
DNaseI hypersensitivity
DNaseI hypersensitivity provides a method to map changes in chromatin structure. The degree of response of the DNA sequence to DNase is classified as generalized sensitivity or hypersensitivity. Generalized nuclease sensitivity is a property inherent in all actively expressed genes (Gazit and Cedar 1980), correlating closely with the presence of acetylated histones. Whether a cause or an effect, the presence of acetylated histones accompanies the appearance of open chromatin extending over 10s to 100s of kilobases and is documented in the chicken lysozyme, chicken ovalbumin, human apolipoprotein B, c-fos, and chicken β-globin loci (Lawson et al. 1982; Goodwin et al. 1985; Jantzen et al. 1986; Feng and Villeponteau 1992; Hebbes et al. 1994). In contrast, hypersensitivity refers to regions showing extreme sensitivity to the cleavage effects of the enzyme that are localized to short stretches of DNA ranging from 100 to 400 bp in length (Gross and Garrard 1988). Extreme sensitivity serves as a marker for functional regions that fall in noncoding sequences; these include promoters, enhancers, silencers, origins of replication, recombination elements, and structural sites of telomeres and centromeres (Cereghini et al. 1984; Gross and Garrard 1988). Early observations suggested that hypersensitivity is associated with the removal of nucleosomes (Almer et al. 1986), whereas more recent analyses can detect the presence of histones in modified form (Gui and Dean 2003), such as acetylated histones H3 and H4 and methylated H3 at lysine 4 (K4; Jenuwein and Allis 2001), at the hypersensitive sites (HSs). The modifications on the histones reduce the affinity of DNA for the nucleosome (Bode et al. 1980), facilitating the interaction of DNA with trans-acting factors (Vettese-Dadey et al. 1996). Thus, the presence of hypersensitivity, which originated as a feature of already-characterized functional sites, has now evolved into a predictive indicator for the presence of a functional site. Furthermore, the impermanent nature of the nuclease hypersensitivity provides insight into the temporal and tissue-specific stages of activity in the underlying elements when assayed using representative biological samples.
Many studies have focused on a locus-specific analysis of nuclease hypersensitive sites. In such studies, the resolution with which one can identify the location of a DNaseI HS varies by approach, ranging from ±500 bp using the indirect end-labeling technique (Wu 1980) to nearly nucleotide resolution using PCR assessment (Yoo et al. 1996) and quantitative PCR (McArthur et al. 2001). Thus, the interpretation of results is dependent on the exact method used for analysis. High-throughput approaches to assess DNase hypersensitivity address the appearance and disappearance of functional sites on a genome-wide scale. Comparisons can be made between cells from different tissues, or within the same type of cell to measure a response to changes in the cellular environment. Two new experimental techniques that have emerged as promising technologies in the ENCODE project (ENCODE Project Consortium 2004) are quantitative chromatin profiling (Dorschner et al. 2004) and massively parallel signature sequencing (Crawford et al. 2005). Additionally, Yuan et al. (2005) have used tiled microarrays to identify translation positioning of nucleosomes in Saccharomyces cerevisiae, revealing that 69% of nucleosomal DNA contained positioned nucleosomes, whereas transcription start sites tended to be nucleosome-free regions. Although the scope and expense of such experiments could limit the pace at which new cell lines or tissue types are investigated, comparisons across samples hold the promise of identifying regions that appear in a specific lineage of cells and thus could provide a systematic means of profiling functional sites that describe a cellular phenotype. Importantly, recent computational advances, which use a sequence-based classification algorithm, have relied on experimental data sets to model hypersensitive sites in silico (Noble et al. 2005). In this approach, a support vector machine is trained to discriminate between experimentally validated HSs and nonHSs. Experimental validation of the genome-wide probability scores shows 70% predictive accuracy, providing support for the extension of this application to additional tissue types. Clearly, additional cycling between experimental validation and computational predictions will continue to improve identification of HSs.
Promoter analyses
Gene expression assays measure changes in the production of a reporter protein in response to cis-acting regulatory signals. For instance, promoter sequences placed upstream of a firefly-luciferase reporter gene (de Wet et al. 1987) or green fluorescent protein (GFP; Tsien 1998) can be introduced into a sample of cultured cells and subsequently assayed in a 24- to 48-h time period, generating reproducible results. Promoters and enhancers can be tested in short-term reactions known as transient transfections, in which the test plasmid remains unintegrated (episomal) in the nucleus. The introduction of an enhancer element creates a “gain-of-function” result, whereas “loss-of-function” assays result from mutations of functional nucleotides in the target region. Alternatively, long-term assays, or stable transfections, use a linearized plasmid that integrates into the genomic DNA and hence is subject to effects conferred by the surrounding chromatin environment. Stable transfections are frequently used to identify sequences that protect against both positive and negative influences of surrounding chromatin (such as boundary elements) and to provide a biologically relevant view of the functional activity as measured within a living cell. High-throughput approaches to cell transfection include the use of cationic lipids or electroporation units that work in a 96-well plate format (Strauss 1996; Ovcharenko et al. 2005; Siemen et al. 2005). One assessment of high-throughput gene expression focused on putative promoters in one percent of the human genome, assayed in multiple cell lines (Trinklein et al. 2003; Cooper et al. 2006). Such large-scale promoter/enhancer assays provide insight into the features commonly found in promoters and serve to verify the functional capability of computationally predicted elements.
The immortalized cell lines used in most experiments rarely recapitulate a “normal” cellular environment (Worton et al. 1977). Nevertheless, they provide a suitable environment in which to initiate studies on the mechanisms of gene regulation rapidly. In contrast, although more technically difficult, in vivo expression assays using animal models supply a means of assessing functional elements within a biologically relevant, tissue-specific context. Analyses using fish, frogs, chickens, and mice (Khokha and Loots 2005; Poulin et al. 2005; Shin et al. 2005; Hallikas et al. 2006; Takemoto et al. 2006) have shown that an element or binding site can act in a defined biological pathway; such conclusions could not have been made with cultured cells. For example, Hallikas et al. (2006) used a computational approach to identify mammalian enhancers and then showed extreme developmental and tissue-specific activity of several of the identified enhancer elements.
Intraspecies comparative approaches in Ciona intestinalis highlight the versatility of this model organism. Boffelli et al. (2004) identified candidate regulatory regions undergoing the slowest mutation rates relative to the surrounding rates and tested them for functional activity in transgenic tadpoles. The work identified a set of noncoding elements that act as tissue-specific enhancers in notochord, endoderm, and neurotube. The availability of genomic sequence from a closely related species, Ciona savignyi, provides opportunities to identify additional candidate regulatory elements through interspecies comparisons. A summary of Web resources and experimental data available for Ciona is provided in Shi et al. (2005).
Protein binding assays
EMSAs and DNaseI protection
Historically, the traditional approach to defining protein–DNA interactions was through the electrophoretic mobility shift assay (EMSA) (Fried and Crothers 1981; Garner and Revzin 1981). By utilizing the sieving power of nondenaturing polyacrylamide gels to separate a protein-bound DNA molecule from one that is unbound, the in vitro “gel-shift” assay is ideal for verifying the ability of an unknown protein to recognize and bind a target DNA sequence. DNaseI protection (also known as DNaseI footprinting) (Galas and Schmitz 1978) is another technique for the precise localization of protein binding sites that does not require knowledge of protein identity. The technique combines the binding reaction of an EMSA with the cleavage reaction of DNaseI. When the radionuclide-labeled probe is visualized on a denaturing polyacrylamide gel, sites protected from cleavage create a blank image in the otherwise semicontinuous ladder of nucleotide positions. As an in vitro assessment of protein binding, DNaseI protection uses a longer probe than EMSA (500 bp vs. 25 bp), elucidating the positions of numerous proteins on the probe simultaneously. A variation of the in vitro assay uses chemical cleavage to produce a uniform cleavage pattern to overcome limitations of the DNaseI enzyme and simplify the interpretation of results (Drouin et al. 2001). With both gel-shift and DNase footprinting assays, unintended DNA–protein interactions are often detected. This can result from interference of nonspecific DNA binding proteins, such as DNA repair proteins, which can bind to the ends of DNA probes in a binding reaction (Klug 1997).
Technical advances related to in vitro binding assays include replacing the use of radionucleotides with use of fluorescent labels (Onizuka et al. 2002) and scaling up for high-throughput approaches. For example, SELEX (systematic evolution of ligands by exponential enrichment) (Tuerk and Gold 1990) and CASTing (cyclic amplification and selection of targets) (Wright et al. 1991) screen large pools of short, random oligonucleotide probes for recognition by a specific protein. The JASPAR database of nonredundant PWMs contains binding site information obtained with this in vitro approach (Sandelin et al. 2004). Other high-throughput in vitro approaches include DIP–ChIP (DNA immunoprecipitation) (Liu et al. 2005) and double-stranded DNA microarray chips (Bulyk et al. 1999; Mukherjee et al. 2004; Bai et al. 2005).
ChIP assays
Use of gel shift assays and in vitro DNase footprinting is quickly giving way to use of assays that capture binding as it happens in the in vivo environment. For instance, the development of in vivo footprinting now allows the study of DNA/protein events within a living cell. This assay uses ligation-mediated PCR (Mueller and Wold 1989) to capture the fractured pieces of genomic DNA that flank the sites protected by protein. Such in vivo assays are informative and can provide tissue-specific information concerning transcription factor binding, yet they can be technically challenging (Komura and Riggs 1998). Also, they do not provide information as to the identity of the involved protein(s). In contrast, the in vivo technique of chromatin immunoprecipitation (ChIP) is especially useful when the protein of interest is known. Reliable protocols for this procedure are listed in Supplemental Table 3. ChIP assays represent a modification of “pull-down” assays in which target proteins are precipitated from solution using an antibody coupled to a retrievable tag. In contrast with standard protein immunoprecipitation assays, ChIP assays capture in vivo protein–DNA interactions by cross-linking proteins to their DNA recognition sites using formaldehyde. Before precipitation by a transcription factor-specific antibody, the DNA is fragmented into small pieces averaging 100–500 bp. After precipitation, reversal of the cross-linking reaction releases the DNA for subsequent detection by PCR amplification. Caveats to the ChIP assay include an inability to detect precise contacts of binding within the 100–500-bp region of the DNA probe and the potential for recovering indirect interactions created by protein–protein contact rather than protein–DNA interactions. Kang et al. (2002) have proposed a method to combine ChIP with DNase protection to address the limitations of both assays, thereby identifying the interacting protein in addition to its interaction site. Although most ChIP assays are performed using tissue culture cells, modifications of the assay have been developed to allow analysis in mammalian tissues (Kirmizis et al. 2003; Chaya and Zaret 2004). A significant challenge that remains for tissue-ChIP assays is in gaining enough tissue for use in the assay, especially if the source tissue is rare (such as for human tumor samples).
ChIP–chip
High-throughput variations of the ChIP technique use ligation-mediated PCR to amplify the pool of DNA sequences as uniformly as possible, generating many copies of all genomic binding sites for a given protein. The assortment of DNA binding sites recovered in a ChIP assay can then be visualized by hybridization to a microarray of genomic sequences. This approach, called ChIP–chip, has been used to interrogate protein–DNA interactions in intact cells (Ren et al. 2000) and is well documented in many comprehensive reviews (see, e.g., Hanlon and Lieb 2004). Just as in cDNA microarrays, DNA that has undergone ChIP assay is labeled with the fluorophore Cy5 and its signal, when bound to an array of target sequences, is compared with signal from an equal amount of total input DNA labeled with Cy3. The relative enrichment of immunoprecipitated DNA over total input DNA (Cy5/Cy3) is used to identify putative binding sites. As the technology improves, the number of searchable target binding sites on the microarray chip grows more complex in nature. For instance, early applications using intergenic regions in yeast (Ren et al. 2000) led the way for analyses of putative promoter regions in humans (Li et al. 2003; Odom et al. 2004). More complex targets were developed using CpG islands associated with promoters (Weinmann and Farnham 2002; Mao et al. 2003; Heisler et al. 2005), finally converging on ever-increasingly refined platforms of nonrepetitive human genomic DNA. A recent study used a series of arrays that contained ∼14 million 50mer oligonucleotides, designed to represent all the non-repeat DNA in the human genome at 100-bp resolution, to define a genome-wide map of active promoters in human fibroblasts (Kim et al. 2005).
One of the key issues in processing the ChIP–chip raw data is to identify the “best” binding sites among the collection of potential DNA targets, pointing to the need for computational scientists to join experimental teams. Several statistical approaches have been developed to detect such regions, which are summarized eloquently in Buck and Lieb (2004). They include a median percentile rank (Lieb et al. 2001), single array-error models (Ren et al. 2000; Li et al. 2003), and a sliding window analysis (Buck and Lieb 2004). The tool ChiPOTle, which uses a sliding window approach, is publicly available for the analysis and interpretation of ChIP–chip data (Buck et al. 2005). Bieda et al. (2006) describe both theoretical and statistical approaches to ChIP–chip data analysis, bringing new insights into the role of the protein E2F1, which acts at a large fraction of human promoters without recognizing a consensus motif. Additional methods are described in a series of recent reports including variance stabilization (Gibbons et al. 2005), enrichment detection (Cawley et al. 2004), and model-based methods (Kim et al. 2005). Programs to identify the most significant regions for protein binding from a ChIP–chip analysis include MPEAK (Kim et al. 2005) and PEAKFinder (Glynn et al. 2004). URLs for these Web sites are listed in Supplemental Table 1.
Experimental conditions and reagent quality greatly affect ChIP–chip results. Published work by Oberley and Farnham (2003) and related papers in the same issue of the journal Methods in Enzymology (Allis and Wu 2003) provide guidance on these issues. Antibodies suitable for ChIP–chip applications are summarized in the ChIP-on-chip database (see Supplemental Table 1), as are additional resources for experimental protocols. ChIP–chip analyses conducted through February 2005 are summarized in Sikder and Kodadek (2005). Repositories and genomic servers listed in Supplemental Table 3 contain additional ChIP–chip data sets, many of which were contributed through the efforts of ENCODE project participants (www.genome.gov/10005107).
Computational follow-up experiments
The ChIP–chip assays described above allow the identification of the genomic region to which a particular protein is bound. However, because of limitations of the assays, it is difficult to identify the exact site within the region to which the protein is bound. Certain computational tools such as MEME (Bailey and Gribskov 1997) and AlignACE (Roth et al. 1998) (see Supplemental Table 1) have proven useful in the follow-up analysis of ChIP–chip data. In addition, other computational approaches have been applied to ChIP–chip data. For example, MDScan (Liu et al. 2002) involves an ab initio motif discovery method and applies a Bayesian statistical score function to refine the candidate motifs enumerated from a set of the top ChIP–chip sequences. Other approaches combine the use of a weight matrix model, which incorporates prior knowledge of PWMs, with statistical classification methods to identify the TFBSs. To illustrate this point, a CART model (classification and regression tree) has been used to identify estrogen receptor α target genes (Jin et al. 2004), and a MARS model (MARSMotifs)—which uses multivariate adaptive regression splines—was selected to discover liver target genes (Smith et al. 2005). Hong et al. (2005) adopted a confidence-rated boosting algorithm to discriminate positive and negative data by taking advantage of the ChIP–chip technology, to distinguish a set of positive data (binding sequences) from a set of negative data (nonbinding sequences). Several studies have increased the sensitivity of motif detection by building motif modules (cis-regulatory modules) based on interacting motifs (Zhou and Wong 2004; Gupta and Liu 2005; Wang et al. 2005; Li et al. 2006). Furthermore, integrating pattern detection of interacting transcription factors, phylogenetic footprinting, and statistical learning methods has provided a substantial increase in the specificity of detecting estrogen receptor alpha (Cheng et al. 2006) and E2F1 target genes (Jin et al. 2006.).
An example of integrating ChIP–chip data with phylogenetic conservation and experimental analyses is shown in Harbison et al. (2004). The authors combined binding data from 203 transcriptional regulators in yeast assayed under more than one growth condition. Six motif discovery methods (including MDScan and MEME) were used to find highly significant motifs for 116 regulatory proteins. The process identified promoter architectures that give clues to regulatory mechanisms defined by the presence of single or repetitive motifs, multiple occurrences of motifs having mixed identities, and co-occurring motifs.
Databases and Web sites for genomic analysis
The databases listed in Supplemental Table 3 serve as repositories for whole-genome high-throughput ChIP–chip binding data. Flexible query and output options in these databases allow one to filter data sets to meet user-specified thresholds (e.g., certain P-values on ChIP–chip data), to pass data to interconnected databases, and to retrieve the DNA sequences that underlie the regions of interest. The Galaxy2 repository (Giardine et al. 2005; Blankenberg et al. 2007) provides mathematical tools, known as set operations, for use on any genomic data sets represented as coordinate-based intervals. These include operations for intersection, union, subtraction, and complement. Importantly, the server can handle extremely large data files. Additional tools include operations for finding all regions that are proximal to a feature data set, merging regions that have overlapping coordinates, and clustering regions that are located within a specified distance. These tools were used to predict MEF2 and MyoD binding sites based on previous knowledge that these sites are known to cluster (Fickett 1996). A complementary approach to predicting target genes from genomic data sets aims to identify target genes using a combination of ChIP–chip and gene expression arrays (Kirmizis and Farnham 2004). Database repositories specializing in both of these data types include GEO (Barrett et al. 2005) and ArrayExpress (Brazma et al. 2003) along with others listed in Supplemental Table 3.
After a set of binding sites and/or target promoters are obtained, further analyses are used to place the information into a wider context. Conservation information is available as alignments of the sequenced mammalian genomes at the UCSC Genome Browser, Vista, and Ensembl Web sites (Karolchik et al. 2003; Frazer et al. 2004; Birney et al. 2006). Additional alignments containing up to 25 mammalian sequences (including pre-eutherian species) are available in the finished ENCODE and ZOOSEQ target regions and are viewable at the Genome Browser. Conversely, stand-alone tools for pairwise or multispecies alignments allow users to create statistically robust alignments of their own target sequences (Brudno et al. 2003; Blanchette et al. 2004; Cooper et al. 2005; for review, see Dubchak and Frazer 2003; Frazer et al. 2003). Highly conserved genomic segments are typically embraced as candidates for experimental and computational predictions of transcription factor binding sites. Accordingly, tools to detect conserved regions, such as Galaxy2, the UCSC Table Browser (Karolchik et al. 2004), MCS Browser (Margulies et al. 2003), and ECR browser (Ovcharenko et al. 2004) allow a user to define and extract conserved sequences from a multi-species alignment. In addition to conservation, the Genome Browser provides predictive measures of regulatory regions such as 5-way regulatory potential (RP) (Kolbe et al. 2004) and PhastCons scores (Siepel et al. 2005), both of which are useful for identifying putative functional regions under selective constraint.
Finally, it is becoming clear that the collection and visualization of data sets comprising both experimental and computational analyses from multiple independent research groups will allow broader insights than individually analyzed data sources (see Supplemental Table 3). Recent studies focused on the experimental and computational analysis of one percent of the human genome (ENCODE Consortium, in prep.) have led to the identification of a large number of novel regulatory elements initially termed GEMMS (genomic elements identified by multiple methods). Visualization of such collected data can be performed using the UCSC browser, which allows the display of information concerning known and predicted genes, protein binding sites, promoter activities, transcription factor motifs, sequence conservation, and DNaseI hypersensitivity sites. Continuing developments focused on the integration and dissemination of combined experimental and computational information are critical for the future.
Conclusions
In this review, we have attempted to demonstrate that the interdependence of experimental and computational approaches allows an iterative refinement process, with each side benefiting from collaboration with the other. Experimentalists may choose to begin a project with in silico analyses or to expand an experimental observation into a genome-wide predictive analysis. Programmers need to verify predictions of binding sites and improve their prognostic pipeline using experimental data. Although many tools for predicting binding sites are available worldwide via the Internet—thereby allowing universal implementation—a lot of experimentalists are not well trained in the programming skills needed for insightful application of the analysis tools. Similarly, despite the availability of robust protocols for genome-scale experimental identification of transcription factor binding sites, these experiments are technically challenging and time consuming. Because programmers are frequently more familiar with the intricacies of tools for binding site prediction, and biologists are better trained in the collection and interpretation of experimental data sets, collaborative interactions and cross training will serve both communities well.
Acknowledgments
We thank the members of our laboratories for productive discussions and anonymous reviewers for helpful comments. We apologize to colleagues whose original contributions could not be cited, given the space constraints. The Intramural Program of the NIH, NHGRI, supports research in the laboratory of L.E.; S.J. is a scholar of the Michael Smith Foundation for Health Research; and P.J.F. and V.X.J. are supported by grants from the NIH (CA45240, HG003129, and DK067889).
References
- ↵C.D. Allis,C. Wu,(2003) Chromatin and chromatin remodeling enzymes, Part A, Vol. 375 (Academic Press).
- ↵A. Almer,H. Rudolph,A. Hinnen,W. Horz,(1986) Removal of positioned nucleosomes from the yeast PHO5 promoter upon PHO5 induction releases additional upstream activating DNA elements. EMBO J. 5:2689–2696.
- ↵Y. Bai,Q. Ge,Q. Liu,T. Li,J. Wang,Z. Lu,(2005) A free-labeled method for DNA-binding protein detection using a double-stranded DNA microarray. J. Nanosci. Nanotechnol. 5:1216–1219.
- ↵T.L. Bailey,C. Elkan,(1995) The value of prior knowledge in discovering motifs with MEME. Proc. Int. Conf. Intell. Syst. Mol. Biol. 3:21–29.
- ↵T.L. Bailey,M. Gribskov,(1997) Score distributions for simultaneous matching to multiple motifs. J. Comput. Biol. 4:45–59.
- ↵T. Barrett,T.O. Suzek,D.B. Troup,S.E. Wilhite,W.C. Ngau,P. Ledoux,D. Rudnev,A.E. Lash,W. Fujibuchi,R. Edgar,(2005) NCBI GEO: Mining millions of expression profiles—database and tools. Nucleic Acids Res. 33:D562–D566.
- ↵M. Bieda,X. Xu,M.A. Singer,R. Green,P.J. Farnham,(2006) Unbiased location analysis of E3F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 16:595–605.
- ↵E. Birney,D. Andrews,M. Caccamo,Y. Chen,L. Clarke,G. Coates,T. Cox,F. Cunningham,V. Curwen,T. Cutts,(2006) Ensembl 2006. Nucleic Acids Res. 34:D556–561.
- ↵M. Blanchette,S. Sinha,(2001) Separating real motifs from their artifacts. Bioinformatics 17:S30–S38.
- ↵M. Blanchette,W.J. Kent,C. Riemer,L. Elnitski,A.F. Smit,K.M. Roskin,R. Baertsch,K. Rosenbloom,H. Clawson,E.D. Green,(2004) Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 14:708–715.
- ↵M. Blanchette,A.R. Bataille,X. Chen,C. Poitras,J. Laganiere,C. Lefebvre,G. Deblois,V. Giguere,V. Ferretti,D. Bergeron,(2006) Genome-wide computational prediction of transcriptional regulatory modules reveals new insights into human gene expression. Genome Res. 16:656–668.
- ↵D. Blankenberg,J. Taylor,I. Schenck,J. He,Y. Zhang,M. Ghent,N. Veeraraghavan,I. Albert,W. Miller,K. Makova,(2007) A framework for collaborative analysis of ENCODE data: Making large-scale analyses biologist-friendly. Genome Res. (in press).
- ↵J. Bode,K. Henco,E. Wingender,(1980) Modulation of the nucleosome structure by histone acetylation. Eur. J. Biochem. 10:143–152.
- ↵D. Boffelli,J. McAuliffe,D. Ovcharenko,K.D. Lewis,I. Ovcharenko,L. Pachter,E.M. Rubin,(2003) Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science 299:1391–1394.
- ↵D. Boffelli,C.V. Weer,L. Weng,K.D. Lewis,M.I. Shoukry,L. Pachter,D.N. Keys,E.M. Rubin,(2004) Intraspecies sequence comparisons for annotating genomes. Genome Res. 14:2406–2411.
- ↵A. Brazma,H. Parkinson,U. Sarkans,M. Shojatalab,J. Vilo,N. Abeygunawardena,E. Holloway,M. Kapushesky,P. Kemmeren,G.G. Lara,(2003) ArrayExpress–a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 31:68–71.
- ↵M. Brudno,C. Do,G. Cooper,M.F. Kim,E. Davydov,NISC Comparative Sequencing ProgramE.D. Green,A. Sidow,S. Batzoglou,(2003) LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13:721–731.
- ↵M.J. Buck,J.D. Lieb,(2004) ChIP–chip: Considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics 83:349–360.
- ↵M.J. Buck,A. Nobel,J. Lieb,(2005) ChIPOTle: A user-friendly tool for the analysis of ChIP–chip data. Genome Biol. 6:R97.
- ↵M.L. Bulyk,E. Gentalen,D.J. Lockhart,G.M. Church,(1999) Quantifying DNA-protein interactions by double-stranded DNA arrays. Nat. Biotechnol. 17:573–577.
- ↵S. Cawley,S. Bekiranov,H.H. Ng,P. Kapranov,E.A. Sekinger,D. Kampa,A. Piccolboni,V. Sementchenko,J. Cheng,A.J. Williams,(2004) Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116:499–509.
- ↵S. Cereghini,S. Saragosti,M. Yaniv,D.H. Hamer,(1984) SV40-α-globulin hybrid minichromosomes. Differences in DNaseI hypersensitivity of promoter and enhancer sequences. Eur. J. Biochem. 144:545–553.
- ↵D. Chaya,K.S. Zaret,(2004) Sequential chromatin immunoprecipitation from animal tissues. Methods Enzymol. 376:361–372.
- ↵A.S.L. Cheng,V.X. Jin,M. Fan,L.T. Smith,S. Liyanarachchi,P.S. Yan,Y.W. Leu,M.W. Chan,C. Plass,K.P. Nephew,(2006) Combinatorial analysis of transcription factor partners reveals recruitment of c-MYC to estrogen receptor α-responsive promoters. Mol. Cell 21:393–404.
- ↵F. Chiaromonte,R.J. Weber,K.M. Roskin,M. Diekhans,W.J. Kent,D. Haussler,(2003) The share of human genomic DNA under selection estimated from human-mouse genomic alignments. Cold Spring Harb. Symp. Quant. Biol. 68:245–254.
- ↵F.S. Collins,(2003) Genome research: The next generation. Cold Spring Harb. Symp. Quant. Biol. 8:49–54.
- ↵G.M. Cooper,E.A. Stone,G. Asimenos,NISC Comparative Sequencing ProgramE.D. Green,S. Batzoglou,A. Sidow,(2005) Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 15:901–913.
- ↵S.J. Cooper,N.D. Trinklein,E.D. Anton,L. Nguyen,R.M. Myers,(2006) Comprehensive analysis of transcriptional promoter structure and function in 1% of the human genome. Genome Res. 16:1–10.
- ↵D. Cora,C. Herrmann,C. Dieterich,F. Di Cunto,P. Provero,M. Caselle,(2005) Ab initio identification of putative human transcription factor binding sites by comparative genomics. BMC Bioinformatics 6:110.
- ↵G.E. Crawford,I.E. Holt,J. Whittle,B.D. Webb,D. Tai,S. Davis,E.H. Margulies,Y. Chen,J.A. Bernat,D. Ginsburg,(2005) Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS) Genome Res. 16:123–131.
- ↵J.R. de Wet,K.V. Wood,M. DeLuca,D.R. Helinski,S. Subramani,(1987) Firefly luciferase gene: Structure and expression in mammalian cells. Mol. Cell. Biol. 7:725–737.
- ↵M.O. Dorschner,M. Hawrylycz,R. Humbert,J.C. Wallace,A. Shafer,J. Kawamoto,J. Mack,R. Hall,J. Goldy,P.J. Sabo,(2004) High-throughput localization of functional elements by quantitative chromatin profiling. Nat. Methods 1:219–225.
- ↵T.A. Down,T.J. Hubbard,(2005) NestedMICA: Sensitive inference of over-represented motifs in nucleic acid sequence. Nucleic Acids Res. 33:1445–1453.
- ↵R. Drouin,J.P. Therrien,M. Angers,S. Ouellet,(2001) In vivo DNA analysis. Methods Mol. Biol. 148:175–219.
- ↵I. Dubchak,K.A. Frazer,(2003) Multi-species sequence comparison: The next frontier in genome annotation. Genome Biol. 4:122.
- ↵ENCODE Project Consortium (2004) The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 306:636–640.
- ↵J. Feng,B. Villeponteau,(1992) High-resolution analysis of c-fos chromatin accessibility using a novel DNase I-PCR assay. Biochim. Biophys. Acta 1130:253–258.
- ↵J.W. Fickett,(1996) Coordinate positioning of MEF2 and myogenin binding sites. Gene 172:19–32.
- ↵K.A. Frazer,L. Elnitski,D.M. Church,I. Dubchak,R.C. Hardison,(2003) Cross-species sequence comparisons: A review of methods and available resources. Genome Res. 13:1–12.
- ↵K.A. Frazer,L. Pachter,A. Poliakov,E.M. Rubin,I. Dubchak,(2004) VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 32:W273–W279.
- ↵M. Fried,D.M. Crothers,(1981) Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res. 9:6505–6525.
- ↵M.C. Frith,Y. Fu,L. Yu,J.F. Chen,U. Hansen,Z. Weng,(2004) Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res. 32:1372–1381.
- ↵S. Gadiraju,C.A. Vyhlidal,J.S. Leeder,P.K. Rogan,(2003) Genome-wide prediction, display and refinement of binding sites with information theory-based models. BMC Bioinformatics 4:38.
- ↵D.J. Galas,A. Schmitz,(1978) DNase footprinting: A simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res. 5:3157–3170.
- ↵M.M. Garner,A. Revzin,(1981) A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: Application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res. 9:3047–3060.
- ↵B. Gazit,H. Cedar,(1980) Nuclease sensitivity of active chromatin. Nucleic Acids Res. 8:5143–5155.
- ↵B. Giardine,C. Riemer,R.C. Hardison,R. Burhans,L. Elnitski,P. Shah,Y. Zhang,D. Blankenberg,I. Albert,J. Taylor,(2005) Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 15:1451–1455.
- ↵F.D. Gibbons,M. Proft,K. Struhl,F.P. Roth,(2005) Chipper: Discovering transcription-factor targets from chromatin immunoprecipitation microarrays using variance stabilization. Genome Biol. 6:R96.
- ↵R.A. Gibbs,G.M. Weinstock,M.L. Metzker,D.M. Muzny,E.J. Sodergren,S. Scherer,G. Scott,D. Steffen,K.C. Worley,P.E. Burch,(2004) Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428:493–521.
- ↵E.F. Glynn,P.C. Megee,H.G. Yu,C. Mistrot,E. Unal,D.E. Koshland,J.L. Gerton,(2004) Genome-wide mapping of the cohesin complex in the yeast Saccharomyces cerevisiae. PLoS Biol. 2:e259.
- ↵G.H. Goodwin,R.H. Nicolas,P.N. Cockerill,S. Zavou,C.A. Wright,(1985) The effect of salt extraction on the structure of transcriptionally active genes; evidence for a DNaseI-sensitive structure which could be dependent on chromatin structure at levels higher than the 30 nm fibre. Nucleic Acids Res. 13:3561–3579.
- ↵D.S. Gross,W.T. Garrard,(1988) Nuclease hypersensitive sites in chromatin. Annu. Rev. Biochem. 57:159–197.
- ↵C.Y. Gui,A. Dean,(2003) A major role for the TATA box in recruitment of chromatin modifying complexes to a globin gene promoter. Proc. Natl. Acad. Sci. 100:7009–7014.
- ↵M. Gupta,J.S. Liu,(2005) De novo cis-regulatory module elicitation for eukaryotic genomes. Proc. Natl. Acad. Sci. 102:7079–7084.
- ↵O. Hallikas,K. Palin,N. Sinjushina,R. Rautiainen,J. Partanen,E. Ukkonen,J. Taipale,(2006) Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell 124:47–59.
- ↵S.E. Hanlon,J.D. Lieb,(2004) Progress and challenges in profiling the dynamics of chromatin and transcription factor binding with DNA microarrays. Curr. Opin. Genet. Dev. 14:697–705.
- ↵M. Harbers,P. Carninci,(2005) Tag-based approaches for transcriptome research and genome annotation. Nat. Methods 7:495–502.
- ↵C.T. Harbison,D.B. Gordon,T.I. Lee,N.J. Rinaldi,K.D. Macisaac,T.W. Danford,N.M. Hannett,J.B. Tagne,D.B. Reynolds,J. Yoo,(2004) Transcriptional regulatory code of a eukaryotic genome. Nature 431:99–104.
- ↵R. Harr,M. Haggstrom,P. Gustafsson,(1983) Search algorithm for pattern match analysis of nucleic acid sequences. Nucleic Acids Res. 11:2943–2957.
- ↵T.R. Hebbes,A.L. Clayton,A.W. Thorne,C. Crane-Robinson,(1994) Core histone hyperacetylation co-maps with generalized DNaseI sensitivity in the chicken β-globin chromosomal domain. EMBO J. 13:1823–1830.
- ↵L.E. Heisler,D. Torti,P.C. Boutros,J. Watson,C. Chan,N. Winegarden,M. Takahashi,P. Yau,H. Huang,P.J. Farnham,(2005) CpG Island microarray probe sequences derived from a physical library are representative of CpG Islands annotated on the human genome. Nucleic Acids Res. 33:2952–2961.
- ↵G.Z. Hertz,G.D. Stormo,(1999) Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 15:563–577.
- ↵P. Hong,X.S. Liu,Q. Zhou,X. Lu,J.S. Liu,W.H. Wong,(2005) A boosting approach for motif modeling using ChIP–chip data. Bioinformatics 21:2636–2643.
- ↵B.R. Huber,M.L. Bulyk,(2006) Meta-analysis discovery of tissue-specific DNA sequence motifs from mammalian gene expression data. BMC Bioinformatics 7:229.
- ↵K. Jantzen,H.P. Fritton,T. Igo-Kemenes,(1986) The DNaseI sensitive domain of the chicken lysozyme gene spans 24 kb. Nucleic Acids Res. 14:6085–6099.
- ↵T. Jenuwein,C.D. Allis,(2001) Translating the histone code. Science 293:1074–1080.
- ↵V.X. Jin,Y.-W. Leu,S. Liyanarachchi,H. Sun,T.H.-M. Huang,R.V. Davuluri,(2004) Identifying estrogen receptor α target genes using integrated computational genomics and chromatin immunoprecipitation microarray. Nucleic Acids Res. 32:6627–6635.
- ↵V.X. Jin,A. Rabinovich,S.L. Squazzo,R. Green,P.J. Farnham,(2006) A computational genomics approach to identify cis-regulatory modules from chromatin immunoprecipitation microarray data—A case study using E2F1. Genome Res. (this issue).
- ↵E. Jolly,C.S. Chin,I. Herskowitz,H. Li,(2005) Genome-wide identification of the regulatory targets of a transcription factor using biochemical characterization and computational genomic analysis. BMC Bioinformatics 6:275.
- ↵S.H. Kang,K. Vieira,J. Bungert,(2002) Combining chromatin immunoprecipitation and DNA footprinting: A novel method to analyze protein-DNA interactions in vivo. Nucleic Acids Res. 30:e44.
- ↵D. Karolchik,R. Baertsch,M. Diekhans,T. Furey,A. Hinrichs,Y. Lu,K.M. Roskin,M. Schwartz,C.W. Sugnet,D.J. Thomas,(2003) The UCSC Genome Browser Database. Nucleic Acids Res. 31:51–54.
- ↵D. Karolchik,A. Hinrichs,T. Furey,K. Roskin,C. Sugnet,D. Haussler,W.J. Kent,(2004) The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32:D493–D496.
- ↵S. Keles,M.J. van der Laan,S. Dudoit,B. Xing,M.B. Eisen,(2003) Supervised detection of regulatory motifs in DNA sequences. Stat. Appl. Genet. Mol. Biol. 2:Article5.
- ↵M.K. Khokha,G.G. Loots,(2005) Strategies for characterising cis-regulatory elements in Xenopus. Brief Funct. Genomic Proteomic. 4:58–68.
- ↵T. Kim,L. Barrera,M. Zheng,C. Qu,M. Singer,T. Richmond,Y. Wu,R.D. Green,B. Ren,(2005) A high-resolution map of active promoters in the human genome. Nature 436:876–878.
- ↵D.C. King,J. Taylor,L. Elnitski,F. Chiaromonte,W. Miller,R.C. Hardison,(2005) Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res. 15:1051–1060.
- ↵A. Kirmizis,P.J. Farnham,(2004) Genomic approaches that aid in the identification of transcription factor target genes. Exp. Biol. Med. 229:705–721.
- ↵A. Kirmizis,S.M. Bartley,P.J. Farnham,(2003) Identification of the polycomb group protein SU(Z)12 as a potential molecular target for human cancer therapy. Mol. Cancer Ther. 2:113–121.
- ↵J. Klug,(1997) Ku autoantigen is a potential major cause of nonspecific bands in electrophoretic mobility shift assays. Biotechniques 22:212–214.
- ↵D. Kolbe,J. Taylor,I.L. Elnitsk,P. Eswara,J. Li,W. Miller,R. Hardison,F. Chiaromonte,(2004) Regulatory potential scores from genome-wide three-way alignments of human, mouse, and rat. Genome Res. 14:700–707.
- ↵J. Komura,A.D. Riggs,(1998) Terminal transferase-dependent PCR: A versatile and sensitive method for in vivo footprinting and detection of DNA adducts. Nucleic Acids Res. 26:1807–1811.
- ↵G. Kreiman,(2004) Identification of sparsely distributed clusters of cis-regulatory elements in sets of co-expressed genes. Nucleic Acids Res. 32:2889–2900.
- ↵W. Krivan,W.W. Wasserman,(2001) A predictive model for regulatory sequences directing liver-specific transcription. Genome Res. 11:1559–1566.
- ↵C.E. Lawrence,S.F. Altschul,M.S. Boguski,J.S. Liu,A.F. Neuwald,J.C. Wootton,(1993) Detecting subtle sequence signals: A Gibbs sampling strategy for multiple alignment. Science 262:208–214.
- ↵G.M. Lawson,B.J. Knoll,C.J. March,S.L. Woo,M.J. Tsai,B.W. O’Malley,(1982) Definition of 5′ and 3′ structural boundaries of the chromatin domain containing the ovalbumin multigene family. J. Biol. Chem. 257:1501–1507.
- ↵M. Levine,R. Tjian,(2003) Transcription regulation and animal diversity. Nature 424:147–151.
- ↵Z. Li,S. Van Calcar,C. Qu,W.K. Cavenee,M.Q. Zhang,B. Ren,(2003) A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc. Natl. Acad. Sci. 100:8164–8169.
- ↵H. Li,H. Chen,L. Bao,K.F. Manly,E.J. Chesler,L. Lu,J. Wang,M. Zhou,R.W. Williams,Y. Cui,(2006) Integrative genetic analysis of transcription modules: Towards filling the gap between genetic loci and inherited traits. Hum. Mol. Genet. 15:481–492.
- ↵J.D. Lieb,X. Liu,D. Botstein,P.O. Brown,(2001) Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat. Genet. 28:327–334.
- ↵X.S. Liu,D.L. Brutlag,J.S. Liu,(2002) An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipation microarray experiments. Nat. Biotechnol. 20:835–839.
- ↵Y. Liu,X.S. Liu,L. Wei,R.B. Altman,S. Batzoglou,(2004) Eukaryotic regulatory element conservation analysis and identification using comparative genomics. Genome Res. 14:451–458.
- ↵X. Liu,D.M. Noll,J.D. Lieb,N.D. Clarke,(2005) DIP-chip: Rapid and accurate determination of DNA-binding specificity. Genome Res. 15:421–427.
- ↵D.M. MacAlpine,S.P. Bell,(2005) A genomic view of eukaryotic DNA replication. Chromosome Res. 13:309–326.
- ↵D.Y. Mao,J.D. Watson,P.S. Yan,D. Barsyte-Lovejoy,F. Khosravi,W.W. Wong,P.J. Farnham,T.H. Huang,L.Z. Penn,(2003) Analysis of Myc bound loci identified by CpG island arrays shows that Max is essential for Myc-dependent repression. Curr. Biol. 13:882–886.
- ↵E.H. Margulies,M. Blanchette,NISC Comparative Sequencing ProgramD. Haussler,E.D. Green,(2003) Identification and characterization of multi-species conserved sequences. Genome Res. 13:2507–2518.
- ↵M. McArthur,S. Gerum,G. Stamatoyannopoulos,(2001) Quantification of DNaseI-sensitivity by real-time PCR: Quantitative analysis of DNaseII-hypersensitivity of the mouse β-globin LCR. Mol. Biol. 313:27–34.
- ↵D.N. Messina,J. Glasscock,W. Gish,M. Lovett,(2004) An ORFeome-based analysis of human transcription factor genes and the construction of a microarray to interrogate their expression. Genome Res. 14:2041–2047.
- ↵W. Miller,K.D. Makova,A. Nekrutenko,R.C. Hardison,(2004) Comparative genomics. Annu. Rev. Genomics Hum. Genet. 5:15–56.
- ↵A. Moses,D. Chiang,M.B. Eisen,(2004) Pacific Symposium on Biocomputing, pp. 324–335, Hawaii, Phylogenetic motif detection by expectation-maximization on evolutionary mixtures.
- ↵P.R. Mueller,B. Wold,(1989) In vivo footprinting of a muscle specific enhancer by ligation mediated PCR. Science 246:780–786.
- ↵S. Mukherjee,M.F. Berger,G. Jona,X.S. Wang,D. Muzzey,M. Snyder,R.A. Young,M.L. Bulyk,(2004) Rapid analysis of the DNA-binding specificities of transcription factors with DNA microarrays. Nat. Genet. 36:1331–1339.
- ↵W.S. Noble,S. Kuehn,R. Thurman,M. Yu,J. Stamatoyannopoulos,(2005) Predicting the in vivo signature of human gene regulatory sequences. Bioinformatics 21:i338–i343.
- ↵M.J. Oberley,P.J. Farnham,(2003) Probing chromatin immunoprecipitates with CpG-island microarrays to identify genomic sites occupied by DNA-binding proteins. Methods Enzymol. 371:577–596.
- ↵D.T. Odom,N. Zizlsperger,D.B. Gordon,G.W. Bell,N.J. Rinaldi,H.L. Murray,T.L. Volkert,J. Schreiber,P.A. Rolfe,D.K. Gifford,(2004) Control of pancreas and liver gene expression by HNF transcription factors. Science 303:1378–1381.
- ↵T. Onizuka,S. Endo,M. Hirano,S. Kanai,H. Akiyama,(2002) Design of a fluorescent electrophoretic mobility shift assay improved for the quantitative and multiple analysis of protein-DNA complexes. Biosci. Biotechnol. Biochem. 66:2732–2734.
- ↵I. Ovcharenko,M.A. Nobrega,G.G. Loots,L. Stubbs,(2004) ECR Browser: A tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 32:W280–W286.
- ↵D. Ovcharenko,R. Jarvis,S. Hunicke-Smith,K. Kelnar,D. Brown,(2005) High-throughput RNAi screening in vitro: From cell lines to primary cells. RNA 11:985–989.
- ↵G. Pavesi,P. Mereghetti,G. Mauri,G. Pesole,(2004) Weeder Web: Discovery of transcription factor binding sites in a set of sequences from co-regulated genes. Nucleic Acids Res. 32:W199–W203.
- ↵J.T. Pedersen,J. Moult,(1996) Genetic algorithms for protein structure prediction. Curr. Opin. Struct. Biol. 6:227–231.
- ↵F. Poulin,M.A. Nobrega,I. Plajzer-Frick,A. Holt,V. Afzal,E.M. Rubin,L.A. Pennacchio,(2005) In vivo characterization of a vertebrate ultraconserved enhancer. Genomics 85:774–781.
- ↵A. Prakash,M. Tompa,(2005) Discovery of regulatory elements in vertebrates through comparative genomics. Nat. Biotechnol. 23:1249–1256.
- ↵J. Qian,N. Esumi,Y. Chen,Q. Wang,I. Chowers,D.J. Zack,(2005) Identification of regulatory targets of tissue-specific transcription factors: Application to retina specific gene regulation. Nucleic Acids Res. 33:3479–3491.
- ↵P. Qiu,(2003) Recent advances in computational promoter analysis in understanding the transcriptional regulatory network. Biochem. Biophys. Res. Commun. 309:495–501.
- ↵B. Ren,F. Robert,J.J. Wyrick,O. Aparicio,E.G. Jennings,I. Simon,J. Zeitlinger,J. Schreiber,N. Hannett,E. Kanin,(2000) Genome-wide location and function of DNA binding proteins. Science 290:2306–2309.
- ↵F.P. Roth,J.D. Hughes,P.W. Estep,G.M. Church,(1998) Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat. Biotechnol. 16:939–945.
- ↵E. Roulet,I. Fisch,T. Junier,P. Bucher,N. Mermod,(1998) Evaluation of computer tools for the prediction of transcription factor binding sites on genomic DNA. In Silico Biol. 1:21–28.
- ↵A. Sandelin,W. Alkema,P. Engstrom,W.W. Wasserman,B. Lenhard,(2004) JASPAR: An open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 32:D91–D94.
- ↵T.D. Schneider,(2000) Evolution of biological information. Nucleic Acids Res. 28:2794–2799.
- ↵E. Schwob,(2004) Flexibility and governance in eukaryotic DNA replication. Curr. Opin. Microbiol. 7:680–690.
- ↵W. Shi,M. Levine,B. Davidson,(2005) Unraveling genomic regulatory networks in the simple chordate, Ciona intestinalis. Genome Res. 15:1668–1674.
- ↵J.T. Shin,J.R. Priest,I. Ovcharenko,A. Ronco,R.K. Moore,C.G. Burns,C.A. MacRae,(2005) Human–zebrafish non-coding conserved elements act in vivo to regulate transcription. Nucleic Acids Res. 33:5437–5445.
- ↵T. Shiraki,S. Kondo,S. Katayama,K. Waki,T. Kasukawa,H. Kawaji,R. Kodzius,A. Watahiki,M. Nakamura,T. Arakawa,(2003) Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. 100:15776–15781.
- ↵R. Siddharthan,E. van Nimwegen,E. Siggia,(2004) RECOMB Satellite Workshop on Regulatory Genomics, PhyloGibbs: Incorporating phylogeny and tracking-based significance assessment in a Gibbs sampler.
- ↵R. Siddharthan,E.D. Siggia,E. van Nimwegen,(2005) PhyloGibbs: A Gibbs sampling motif finder that incorporates phylogeny. PLoS Comput. Biol. 1:e67.
- ↵H. Siemen,M. Nix,E. Endl,P. Koch,J. Itskovitz-Eldor,O. Brustle,(2005) Nucleofection of human embryonic stem cells. Stem Cells Dev. 14:378–383.
- ↵A. Siepel,G. Bejerano,J.S. Pedersen,A.S. Hinrichs,M. Hou,K. Rosenbloom,H. Clawson,J. Spieth,L.W. Hillier,S. Richards,(2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15:1034–1050.
- ↵D. Sikder,T. Kodadek,(2005) Genomic studies of transcription factor-DNA interactions. Curr. Opin. Chem. Biol. 9:38–45.
- ↵S. Sinha,E. van Nimwegen,E. Siggia,(2003) Proceedings of the Eleventh International Conference on Intelligent Systems for Molecular Biology, A probabilistic method to detect regulatory modules, (Brisbane, Australia), pp 292–301.
- ↵A.D. Smith,P. Sumazin,D. Das,M.Q. Zhang,(2005) Mining ChIP–chip data for transcription factor and cofactor binding sites. Bioinformatics 21:i403–i412.
- ↵G.D. Stormo,T.D. Schneider,L. Gold,A. Ehrenfeucht,(1982) Use of the ‘Perceptron’ algorithm to distinguish translational initiation sites in E. coli. Nucleic Acids Res. 10:2997–3011.
- ↵W.M. Strauss,(1996) Transfection of mammalian cells via lipofection. Methods Mol. Biol. 54:307–327.
- ↵Y. Suzuki,R. Yamashita,K. Nakai,S. Sugano,(2002) DBTSS: DataBase of human Transcriptional Start Sites and full-length cDNAs. Nucleic Acids Res. 30:328–331.
- ↵D.A. Tagle,B.F. Koop,M. Goodman,J.L. Slightom,D.L. Hess,R.T. Jones,(1988) Embryonic ε and γ globin genes of a prosimian primate (Galago crassicaudatus) J. Mol. Biol. 203:439–455.
- ↵T. Takemoto,M. Uchikawa,Y. Kamachi,H. Kondoh,(2006) Convergence of Wnt and FGF signals in the genesis of posterior neural plate through activation of the Sox2 enhancer N-1. Development 133:297–306.
- ↵M. Tompa,N. Li,T.L. Bailey,G.M. Church,B. De Moor,E. Eskin,A.V. Favorov,M.C. Frith,Y. Fu,W.J. Kent,(2005) Assessing computational tools for the discovery of transcription factor binding sites. Nat. Biotechnol. 23:137–144.
- ↵N.D. Trinklein,S.J. Aldred,A.J. Saldanha,R.M. Myers,(2003) Identification and functional analysis of human transcriptional promoters. Genome Res. 13:308–312.
- ↵R.Y. Tsien,(1998) The green fluorescent protein. Annu. Rev. Biochem. 67:509–544.
- ↵C. Tuerk,L. Gold,(1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249:505–510.
- ↵J. van Helden,(2003) Regulatory sequence analysis tools. Nucleic Acids Res. 31:3593–3596.
- ↵J. van Helden,A.F. Rios,J. Collado-Vides,(2000) Discovering regulatory elements in non-coding sequences by analysis of spaced dyads. Nucleic Acids Res. 28:1808–1818.
- ↵T. Vavouri,G. Elgar,(2005) Prediction of cis-regulatory elements using binding site matrices—the successes, the failures and the reasons for both. Curr. Opin. Genet. Dev. 15:395–402.
- ↵M. Vettese-Dadey,P.A. Grant,T.R. Hebbes,C. Crane-Robinson,C.D. Allis,J.L. Workman,(1996) Acetylation of histone H4 plays a primary role in enhancing transcription factor binding to nucleosomal DNA in vitro. EMBO J. 15:2508–2518.
- ↵C.A. Vyhlidal,P.K. Rogan,J.S. Leeder,(2004) Development and refinement of pregnane X receptor (PXR) DNA binding site model using information theory: Insights into PXR-mediated gene regulation. J. Biol. Chem. 279:46779–46786.
- ↵T. Wang,G.D. Stormo,(2003) Combining phylogenetic data with co-regulated genes to identify regulatory motifs. Bioinformatics 19:2369–2380.
- ↵W. Wang,J.M. Cherry,Y. Nochomovitz,E. Jolly,D. Botstein,H. Li,(2005) Inference of combinatorial regulation in yeast transcriptional networks: A case study of sporulation. Proc. Natl. Acad. Sci. 102:1998–2003.
- ↵W.W. Wasserman,J.W. Fickett,(1998) Identification of regulatory regions which confer muscle-specific gene expression. J. Mol. Biol. 278:167–181.
- ↵R.H. Waterston,K. Lindblad-Toh,E. Birney,J. Rogers,J.F. Abril,P. Agarwal,R. Agarwala,R. Ainscough,M. Alexandersson,P. An,(2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562.
- ↵A.S. Weinmann,P.J. Farnham,(2002) Identification of unknown target genes of human transcription factors using chromatin immunoprecipitation. Methods 26:37–47.
- ↵S. Weisbrod,H. Weintraub,(1979) Isolation of a subclass of nuclear proteins responsible for conferring a DNaseI-sensitive structure on globin chromatin. Proc. Natl. Acad. Sci. 76:630–634.
- ↵R.G. Worton,C.C. Ho,C. Duff,(1977) Chromosome stability in CHO cells. Somatic Cell Genet. 3:27–45.
- ↵W.E. Wright,M. Binder,W. Funk,(1991) Cyclic amplification and selection of targets (CASTing) for the myogenin consensus binding site. Mol. Cell. Biol. 11:4104–4110.
- ↵C. Wu,(1980) The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNaseI. Nature 286:854–860.
- ↵X. Xie,J. Lu,E.J. Kulbokas,T.R. Golub,V. Mootha,K. Lindblad-Toh,E.S. Lander,M. Kellis,(2005) Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434:338–345.
- ↵J. Yoo,L.E. Herman,C. Li,S.B. Krantz,D. Tuan,(1996) Dynamic changes in the locus control region of erythroid progenitor cells demonstrated by polymerase chain reaction. Blood 87:2558–2567.
- ↵G.C. Yuan,Y.J. Liu,M.F. Dion,M.D. Slack,L.F. Wu,S.J. Altschuler,O.J. Rando,(2005) Genome-scale identification of nucleosome positions in S. cerevisiae. Science 309:626–630.
- ↵Q. Zhou,W.H. Wong,(2004) CisModule: De novo discovery of cis-regulatory modules by hierarchical mixture modeling. Proc. Natl. Acad. Sci. 101:12114–12119.
- ↵Z. Zhu,J. Shendure,G.M. Church,(2005) Discovering functional transcription-factor combinations in the human cell cycle. Genome Res. 15:848–855.
Notes
[1] [Supplemental material is available online at www.genome.org.]
[2] Article published online before print. Article and publication date at http://www.genome.org/cgi/doi/10.1101/gr.4140006