Abstract
Chromosome-specific low-copy repeats, or duplicons, occur in multiple regions of the human genome. Homologous recombination between different duplicon copies leads to chromosomal rearrangements, such as deletions, duplications, inversions, and inverted duplications, depending on the orientation of the recombining duplicons. When such rearrangements cause dosage imbalance of a developmentally important gene(s), genetic diseases now termed genomic disorders result, at a frequency of 0.7–1/1000 births. Duplicons can have simple or very complex structures, with variation in copy number from 2 to >10 repeats, and each varying in size from a few kilobases in length to hundreds of kilobases. Analysis of the different duplicons involved in human genomic disorders identifies features that may predispose to recombination, including large size and high sequence identity between the recombining copies, putative recombination promoting features, and the presence of multiple genes/pseudogenes that may include genes expressed in germ cells. Most of the chromosome rearrangements involve duplicons near pericentromeric regions, which may relate to the propensity of such regions to accumulate duplicons. Detailed analyses of the structure, polymorphic variation, and mechanisms of recombination in genomic disorders, as well as the evolutionary origin of various duplicons will further our understanding of the structure, function, and fluidity of the human genome.
Although the primary cause of many genetic diseases is single gene defects, a number of syndromes have been defined as genomic disorders (Lupski 1998), as the majority of cases arise from structural rearrangements of certain regions of a chromosome. In such cases, disease is due to dosage imbalance of one or more genes, caused by chromosomal structural change and not by mutation of the gene(s) itself. Rather than the disease etiology, this review focuses on the structural mechanisms of chromosome rearrangements.
Structural changes of certain chromosomal segments were first associated with several human genetic disorders in the early 1980s, such as Prader-Willi syndrome at 15q11–q13 (Ledbetter et al. 1981) and DiGeorge syndrome at 22q11 (de la Chapelle et al 1981). Different terms (e.g., contiguous gene, microdeletion, contiguous deletion, and segmental aneusomy) were given to describe these and other syndromes later associated with a specific structural change of a chromosome, including the more apt term, genomic disorder (Lupski 1998). Whereas the term genomic disorder (Lupski 1998) includes all syndromes involving a change at the genomic level, the following discussion is restricted to genomic disorders arising by homologous recombination between region-specific, low-copy repeats during meiosis. Recent studies show that duplications of small chromosome segments may not be a rare event. Analysis of long stretches of available sequence from human chromosomes 22 and X suggest that 5%–10% of the genome may be duplicated (Mazzarella and Schlessinger 1998; Dunham et al. 1999). Sequence homology between duplicated DNA segments provides a chance for misalignment during meiosis, leading to unequal exchange and chromosome rearrangement, by either inter- or intrachromosomal or sister chromatid homologous recombination (Fig. 1). This process with divergence between duplicated segments is essential to the generation of diversity and new genes over evolutionary time, although the more typical, short-term effect is genetic disease.
Genomic rearrangements resulting from recombination between duplicons. (a) Interchromosomal recombination between direct repeats results in deletion and/or duplication. (b) Intrachromosomal recombination between direct repeats results in deletion. (c) Intrachromosomal recombination between inverted repeats results in an inversion. Repeated sequences are depicted as yellow arrows with the orientation indicated by the direction of the arrows. Recombination is shown by the red X. Upper- and lowercase letters refer to both alleles of the flanking unique sequence (e.g., A and a). Adapted in part fromLupski (1998).

Region-specific, low-copy repeat elements, or duplicons (Eichler 1998), have been identified flanking many regions susceptible to human genomic disorders (for recent reviews, see Purandare and Patel 1997; Lupski 1998; Mazzarella and Schlessinger 1998). These disorders arise from chromosomal rearrangements mediated by homologous recombination between different copies of a duplicon with deletions (Fig. 1a,b), duplications (Fig. 1a), or inversions (Fig. 1c), the outcome depending on unknown factors, although the orientation of duplicons is clearly one important criterion. Characterization of various genomic disorders showed that they can be classified into three groups. In the first group, the length of the repeated segments and the distance between the repeat units are both small. The duplicon in such cases is either a gene cluster or repeated sequences flanking a gene. Homologous recombination between the duplicons generates an imbalance of a dosage-sensitive gene, leading to a disease phenotype. This group includes a number of disorders such as red-green color blindness and α- or β-thalassemia (Lupski 1998), and will not be discussed further here. The other two groups of genomic disorders involve rearrangement of a large segment of the chromosome, yet the flanking duplicons are structurally simple in the second group, whereas the third group of disorders involve complex duplicons. In both cases, if the rearrangement is a deletion or duplication, the expression level of multiple genes may be affected, although the phenotypes can arise from dosage imbalance of a single gene. The affected gene(s) may or may not be part of the duplicon, and can thus be situated quite far from the repeats.
Simple Duplicons Mediating Genomic Disorders
S232 Elements and X-Linked Ichthyosis
Deletions of the steroid sulfatase (STS) gene in males cause a skin disorder, X-linked ichthyosis. The incidence of this disorder is 1 in 2–5000 males, with ∼90% of the patients having a deletion of 1.9 Mb that includes the STS locus. The deletions are mediated by homologous recombination between duplicons called S232 elements, situated at either end of the deletion (Ballabio et al 1990;Yen et al. 1990). There are 6–14 S232 duplicons, 4–12 in Xp22.3 and 2 on the Y chromosome, and each is composed of a 5-kb unique sequence and two VNTR (variable-number tandem repeat) sequences (Li et al. 1992;Lahn and Page 2000). One mapped S232 duplicon lies distal to theSTS locus and three proximal to STS, with recombination proposed to occur between the former and only one of the latter in tandem (Fig. 2a; Li et al 1992). No cases involve recombination with the Y chromosome (Yen et al 1990). Recent studies show that a copy of the VCX gene is present within the S232 duplicon, and that the VCX gene family is expressed exclusively in male germ cells (Lahn and Page 2000).
Genes and duplicons in genomic disorders involving simple duplicons. (a) Xp22.32 deletions in X-linked icthyosis; (b) reciprocal 17p12 duplications in Charcot-Marie Tooth syndrome 1A (CMT1A) and deletions in hereditary neuropathy with liability to pressure palsies (HNPP), (c) Xq28 inversions in hemophilia A; (d) 17q11.2 deletions in neurofibromatosis type 1 (NF1). Duplicons are shown as either triangles (when the orientation is known) or squares (when the orientation is unknown). Unequal crossing-over occurs primarily between duplicon copies depicted by shaded triangles. Percentages reflect the frequency of breakpoints at two alternative sites. Major genes responsible for each syndrome are shown as hatched rectangles. (cen) Centromere; (tel) telomere. (See text for references.)

CMT1A–REP and Two Distinct Peripheral Neuropathies
Reciprocal chromosome rearrangements of 17p12 occur in Charcot-Marie-Tooth syndrome type 1A (CMT1A) and hereditary neuropathy with liability to pressure palsies (HNPP) (Chance and Fischbeck 1994;Patel and Lupski 1994). A 1.5-Mb tandem duplication of 17p12 occurs in 70% of inherited and 90% of sporadic CMT1A cases (Hoogendijk et al. 1992; Wise et al. 1993; Nelis et al. 1996), in which 10%–18% of CMT1A cases overall are de novo (Blair et al. 1996; Bort et al. 1997). A deletion of the same chromosomal segment occurs in 82% of familial and 86% of sporadic HNPP cases (Nelis et al. 1996). The protein encoded by the PMP22, or peripheral myelin protein-22, gene in the 1.5-Mb region appears to be sensitive to subtle dosage changes and is responsible for both syndromes (for review, see Chance and Fischbeck 1994; Patel and Lupski 1994).
The 1.5-Mb region is flanked by a duplicon termed CMT1A–REP (Fig. 2b;Pentao et al. 1992; Chance et al. 1993), with 98.7% sequence identity across 24,011 bp (Reiter et al. 1997). The COX10 gene encoding heme A:farnesyltransferase overlaps the distal duplicon, and a duplicated internal segment of COX10 forms the proximal CMT1A–REP (Reiter et al. 1997). Homologous recombination between these repeat elements lead to the duplications in CMT1A and deletions in HNPP (Lupski 1998). In both CMT1A and HNPP rearrangements, the majority of crossovers happen within a single recombination hot spot (Kiyosawa et al. 1995; Kiyosawa and Chance 1996; Lopes et al. 1996, 1998; Reiter et al. 1996, 1998). Because all crossovers occur in long stretches of identity, the minimum efficient processing segment (MEPS) for efficient meiotic homologous recombination was proposed to be between 337 and 456 bp long. This is similar to other observations of an ∼200 bp MEPS for homologous recombination in mammalian cells (Waldman and Liskay 1988). The finding of a mariner transposon-like element with its 3′ end 1774 bp from the hot spot led to a hypothesis that it could mediate strand exchange events via double-strand breaks induced by a transposase at or near the 3′ end of the element (Reiter et al. 1996, 1998). Multiple sequences similar to human minisatellite consensus sequences [4 copies similar to 5′ -GGGCAGGA(A/G)G-3′, and one copy similar to 5′ -AGAGGTGGG CAGGTGG-3′] and a χ-like sequence (5′ -GCTGGTGG-3′) were also identified in a 270-bp region within the hot spot (Lopes et al. 1998), and suggested to promote homologous recombination events. However, no direct evidence supports the involvement in recombination of a mariner or other specific sequences.
Interestingly, most de novo CMT1A duplications contain both paternal haplotypes and therefore arose from unequal crossing over involving both homologs (Palau et al 1993; Lopes et al., 1997,1998). In contrast, all four maternal cases involving CMT1A–REP recombination (two duplications and two deletions) are intrachromosomal events (Lopes et al. 1997), which suggests a mechanistic difference during spermatogenesis and oogenesis for 17p12 rearrangements. Using themariner-like sequence as a probe, a 2.2-kb transcript was claimed to be present in testis but not in ovary (Kiyosawa and Chance 1996), although the genomic source(s) of the putative transcripts are unknown and the data is not convincing. Nevertheless, an open chromatin structure due to transcription of COX10 or themariner could be associated with generation of CMT1A duplications during spermatogenesis.
int22h Elements and Severe Hemophilia A
Hemophilia A is an X-linked coagulation disorder affecting ∼1 in 5000 males (Soucie et al. 1998), due to mutations of the factor VIII gene (F8). About 45% of severe hemophilia A males carry a large (∼500 kb) inversion on Xq, which disrupts F8 (Lakich et al. 1993; Naylor et al. 1993). Most inversions are intrachromosomal (Lakich et al. 1993) and originate almost exclusively in male germ cells (Rossiter et al. 1994). The inversion occurs by homologous recombination between 9.5-kb duplicons (int22h, or intron 22 homologous region), one in intron 22 of F8, and either of two homologs oriented in the opposite direction and located 500 kb more distal (Fig. 2c; Naylor et al. 1995). In one case, the intron 22 copy recombined with a fourth, truncated copy of int22h, to generate a smaller inversion (Naylor et al. 1996). An 8-kb segment ofint22h is 99.9% similar among the three copies, with a 1.8-kb intronless gene (F8A) within each of the repeat elements (Levinson et al. 1990), although it is not known whether F8Ais expressed during spermatogenesis.
Germ-line Deletions of 17q11.2 in Neurofibromatosis Type 1
Recent studies indicate that a minority (2%–13%) of neurofibromatosis type l (NF1) cases result from an NF1 gene deletion of 1.5 Mb that is mediated by homologous recombination between duplicons termed NF1REPs (Dorschner et al. 2000). These duplicons are direct repeats that span 15–100 kb and contain several genes/pseudogenes (a third copy maps distally in 17q24). Of theNF1 deletions that were de novo, 75% were maternal in origin, although somatic mosaicism for NF1 deletions have been described suggesting that mitotic recombination between NF1REPs may also occur (Dorschner et al. 2000).
Genomic Disorders Arising from Chromosome Rearrangements Involving Complex Duplicons
Recent molecular and cytological studies have identified large, complex duplicons in the breakpoint regions of several human genomic disorders. The rearranged DNA segments usually span several megabases and are flanked by large duplicons that often contain several genes and/or pseudogenes. Multiple copies are present both at the common breakpoint regions and elsewhere within the same subchromosomal region. On the basis of limited analyses, sequence identity between different duplicon copies are usually >90% and up to 99%. As the duplicons are large in size, have high homology, and multiple copies, the characterization of these duplicons has proven to be difficult. With the recent development of long-range mapping and sequencing techniques, the nature of some of these rearrangements has begun to emerge.
Chromosome 7q11.23 Deletions and Williams syndrome
Williams syndrome (WS) is a multisystem developmental disorder affecting connective tissue and the central nervous system, with an estimated incidence of 1 in 20,000 live births (Morris et al. 1993;Sadler et al. 1993). Most (95%) patients have a de novo ∼1.6-Mb deletion of chromosome 7q11.23, including the elastin(ELN) gene (Ewart et al. 1993; Nickerson et al. 1995), with common deletion breakpoints (Perez Jurado et al. 1996; Robinson et al. 1996). The deletions arise equally often from both paternal and maternal meiosis, although a potential role for genomic imprinting in clinical features of WS (Perez Jurado et al. 1996) remains controversial (Francke 1999; Wang et al. 1999).
Duplicons of 200–300 kb that flank the common WS deletion (Fig.3a; Francke 1999) each contain up to three transcripts, including at least three duplicons that have sequences related to the PMS2 mismatch repair gene (Osborne et al. 1997). Two of the duplicons contain copies of the transcription factorGTF2I (Perez Jurado et al. 1998), in which the distal copy is the ancestral GTF2I gene with a unique 5′ end within the common deletions, whereas the proximal copy represents a truncated, expressed pseudogene (GTF2IP1). The neutrophil cytosolic factor 1 (NCF1) gene maps telomeric to GTF2I in the distal duplicon, whereas an NCF1 pseudogene is present at both the proximal and the distal duplicons (Hockenhull et al. 1999). A shared >3-Mb junction NotI fragment in WS deletions indicates clustering of breakpoints (Perez Jurado et al. 1998), suggesting that homologous recombination between the duplicons is responsible for generating the WS deletion, with an interchromosomal event in most cases (Dutly and Schinzel 1996; Baumer et al. 1998).
Genes and duplicons in genomic disorders involving complex duplicons. (a) 7q11.23 deletions in WS; (b) reciprocal 17p11.2 deletions in SMS and duplications in duplication 17p11.2 syndrome; (c) 22q11 deletions in DiGeorge and VCFS syndromes; (d) common deletions in the imprinted PWS and AS. Duplicons, unequal crossing-over positions, as well as frequency, genes, and abbreviations, are denoted as in Fig. 2. Triangles pointing in both directions reflect the complex modules revealed by 22q11 sequence. A major gene contributing to Williams syndrome,elastin, is shown as a hatched rectangle, as is theUBE3A gene involved in Angelman syndrome (AS). Genes that contribute to Smith-Magenis, duplication 17p11.2, DiGeorge/VCFS, and Prader-Willi syndromes are unknown. (See text for references.)

Chromosome 17p11.2 Deletions (SMS) and Duplications
Smith-Magenis syndrome (SMS) is a mental retardation/multiple congenital anomalies syndrome that occurs in 1/25,000 births (Greenberg et al. 1991). Most patients (>90%) have an ∼5-Mb common deletion of 17p11.2 (Juyal et al. 1996; Chen et al. 1997; Lupski 1998). A cluster of four genes/ pseudogenes (SRP, TRE, KER, CLP) spanning >200 kb was found to be duplicated and was mapped to the common SMS deletion breakpoint regions (Chen et al. 1997), with a third copy mapping within the 5-Mb deleted region (Fig. 3b). The gene CLP (for coactosin-like protein) maps only to the proximal (P) and distal (D) breakpoint regions (SMS–REPP and D), whereas the other three genes also map to the middle duplicon (SMS–REPM). Additional copies of CLPoccur in chromosomes 15, 16, and Y, but are uncharacterized. A novelNotI junction fragment of 1.2 Mb is identified, using aCLP cDNA probe, in every SMS patient with the common deletion, suggesting recombination between the outer SMS–REP duplicons (Chen et al. 1997). No chromosome rearrangement involving the middle copy of SMS–REP has been identified to date. Interestingly, a 210 bp sequence in the 3′ UTR of CLP has >95% identity to a human cDNA sequence with autonomously replicating activity (Wu et al. 1993). Initiation of DNA replication from the latter element, or transcription through the CLP region, may be sufficient to unwind DNA and render the region prone to homologous recombination (Chen et al. 1997).
Several 17p11.2 duplication patients have been identified, with mild mental retardation and minor dysmorphism (Chen et al 1997; Potocki et al 2000). Using the CLP cDNA probe, a novel ∼1.1-MbNotI junction fragment was detected in the duplication patients. This result and other molecular studies suggested that the 17p11.2 duplications involve the SMS–REP and represent reciprocal interchromosomal recombination events to those in SMS (Chen et al. 1997; Potocki et al. 2000). The 17p11.2 duplications were de novo and preferentially paternal in origin (Potocki et al. 2000).
Low Copy Repeats and Chromosome 22q11 Rearrangements
Several congenital anomaly disorders are associated with de novo germ-line rearrangements of 22q11 that involve duplicon-mediated homologous recombination (Edelmann et al. 1999 a,b,c; Shaikh et al. 2000). DiGeorge and velocardiofacial syndromes (VCFS) are the most common microdeletion syndrome, with an incidence of 1 in 4000 births (Wilson et al. 1993). The cardiac and craniofacial features of VCFS may result from haploinsufficiency for the UFD1L gene (Yamagishi et al. 1999), although this remains controversial (Baldini 1999). Cat-eye syndrome (CES) is a rare multiple anomalies disorder (Schinzel et al. 1981) associated with a dicentric bisatellited supernumerary chromosome 22 (pter>q11.2∷q11.2->pter). Derivative 22 [der(22)] syndrome is a rare mental retardation/multiple anomaly disorder, due to partial trisomy of chromosome 22q11 by unbalanced segregation of a translocation (Funke et al. 1999; Edelmann et al. 1999c).
About 90% of VCFS deletion patients have a similar deletion of 3 Mb, whereas 7% of patients have a 1.5-Mb deletion with the same proximal breakpoint region as in the 3-Mb deletions (Carlson et al. 1997;Edelmann et al. 1999b; Shaikh et al. 2000). A low-copy repeat (LCR22) ranging from 40 to 350 kb and 97% to 98% identity occurs in the vicinity of all three 22q11 breakpoint regions, as well as at six adjacent locations over a 6.5-Mb region (Fig. 3c; Dunham et al. 1999;Shaikh et al. 2000). Each LCR22 duplicon differs in content and organization of complex sequence modules (Dunham et al. 1999; Shaikh et al. 2000), including variable content and arrangements of 11 genes/ pseudogenes (GGT, BCRL, V7-Rel, HMPLPL, GGT-REL, E2F6L, ISG43, KIAA0049, DGCR6, PRODH, NF1L). Several potential recombination sequences within the LCR22 duplicons include VNTR sequences and AT-rich repeats (Edelmann et al. 1999c;Shaikh et al. 2000). Most recombination events appear to occur within the LCR22 repeats (Edelmann et al. 1999a b c; Funke et al. 1999; Shaikh et al. 2000). There is no bias for the parental origin of 22q11 deletions leading to VCFS, with both inter- and intra-chromosomal events observed (Baumer et al. 1998; Edelmann et al. 1999a).
Interestingly, misalignment of different LCR22s during meiosis may generate both deletions and reciprocal duplications; an interstitial duplication of the same 3-Mb DNA segment as in the VCFS deletion has been identified in one family (Edelmann et al. 1999b). Two CES breakpoint clusters also map to the LCR22 duplicons associated with the proximal and distal breakpoint regions of the common VCFS deletions (McTaggart et al. 1998). Der (22) chromosomes break within the nested middle copy of LCR22 (Edelmann et al. 1999b, c; Funke et al. 1999). Other LCR22 duplicons may also be used in generating rare chromosomal rearrangements, including somatic rearrangements that lead to malignant diseases (Edelmann et al. 1999b). The identification and sequence analysis of exact breakpoints in patients with these diverse rearrangements of 22q11.2 will shed significant light on the molecular events that predispose to recombination within complex modular duplicons.
END Repeats and Chromosome 15q11–15q13 Deletions
Proximal chromosome 15q is also involved in an astonishing variety of cytogenetic aberrations with a high frequency. Prader-Willi and Angelman syndromes (PWS and AS) occur at a frequency of ∼1 in 15, 000 births, and most cases are due to an ∼4-Mb deletion of 15q11–q13 of paternal or maternal origin, respectively (Nicholls et al. 1998; Jiang et al. 1999). PWS and AS are models for genomic imprinting in human (Nicholls et al. 1998), although this is not considered further here. The vast majority (>95%) of the deletion patients have clustered deletion breakpoint regions, with a single distal breakpoint and two proximal breakpoint regions (Christian et al. 1995; Amos-Landgraf et al. 1999). There is no apparent mechanistic differences for paternal and maternal deletions, as both inter- and intra-chromosomal rearrangements are seen in PWS and AS patients (Carrozzo et al. 1997; Robinson et al. 1998).
Recent molecular and cytogenetic analyses demonstrated at least 10 copies of a complex duplicon, termed the END repeat, that maps to the three common deletion breakpoint regions, with two chromosome 16p11 copies (Fig. 3d; Buiting et al. 1992, 1998; Amos-Landgraf et al. 1999; Ji et al. 1999). The END repeats are composed to a large extent of duplications of a huge gene, HERC2, although multiple rearrangements within the derived duplicons have occurred during evolution (Amos-Landgraf et al. 1999; Ji et al. 1999, 2000). The ancestral HERC2 gene encodes a giant protein, recessive mutations of which cause a neurological and developmental syndrome in mouse (Ji et al. 1999). A simple model to explain the generation of PWS and AS common deletions proposes homologous misalignment and meiotic recombination between different END repeat copies in proximal and distal 15q11–q13 (Amos-Landgraf et al. 1999). Active transcription of HERC2 and related pseudogenes in male and female germ-line tissues suggest that they are transcribed in germ cells (Amos-Landgraf et al. 1999), which is supported by jdf2 mouse phenotypic studies (Ji et al. 1999), and thus the resultant open chromatin structure may further stimulate recombination. Identification of a putative expressed breakpoint junction (using a HERC2 probe) in one PWS patient of only five tested supports this model (Amos-Landgraf et al. 1999).
Christian et al. (1999) independently identified duplicons that map to the breakpoint regions of PWS/AS common deletions, by STS content mapping of genomic clones at or near the proximal and distal PWS/AS breakpoints. Several STSs and ESTs were mapped to duplicons at breakpoints 2 and 3 (BP2 and BP3). Interphase FISH using a PAC clone containing duplicon sequences identified ∼3 signals in 15q11–q13 from normal chromosomes, and a reduced number of signals in deletion chromosomes (as well as signals on 16p11 and 15q24; Christian et al. 1999). However, the difficulty in distinguishing closely mapping, highly homologous, and partial (truncated) sequences resulted in an underestimation of the number of copies of the duplicon.
One end of the END-repeat duplicons is within the 3′ part of HERC2 (Ji et al. 1999). The HERC2-related content of different duplicons is 36.6 kb in the most rearranged/deleted copy in 16p11.2 to an estimated ∼150–200 kb in some of the chromosome 15 duplicons (Ji et al. 2000). Nevertheless, the end point of the duplicon 5′ of HERC2 is unknown. Additional genes are present in the duplicons, including the smallMYLE gene (Christian et al. 1999), although only four of the seven potential transcripts identified may represent new genes/pseudogenes in the END repeats (discussed in Ji et al. 2000). Furthermore, there is evidence that independent duplicons flank the HERC2 (and MYLE)-containing duplicons in 15q11.2 (Buiting et al. 1999). Additional experiments, particularly complete genomic sequence, are needed to further characterize the content, orientation, and copy number of the END repeat duplicons that may be involved in the mechanism for PWS/AS common deletions.
Other 15q11–15q13 Rearrangements
The most common of these involve marker chromosomes. FISH studies indicate that ∼57% of all marker chromosomes in newborns originate from chromosome 15, with most an inv dup(15), that occur in ∼ 0.4/1000 births (Blennow et al. 1995). Inv dup(15) chromosomes are bisatellited chromosomes derived from chromosome 15, which contain two centromeric regions, although usually only one centromere is active (Schwartz and Depinet 1996). The inv dup(15) is a mirror image chromosome that is duplicated about a central axis (e.g., pter—cen—q13—cen—pter). It is a misnomer to call it an inv dup(15), and preferred terms would be either a derivative or dicentric chromosome; however, it is normally referred to as an inv dup(15) because of historical context. These chromosomes come in a variety of size classes. Small marker chromosomes either have only one centromere or one of the two proximal breakpoints associated with PWS and AS; that is, for type 1, a breakpoint between the centromere (D15Z1) and markers located between BP1 and BP2, or for type 2, a breakpoint within the BP2 region (Cheng et al. 1994; Huang et al. 1997). Larger marker chromosomes can have variable breakpoints (Mignon et al. 1996), although most either share the distal PWS/AS breakpoint (the BP3 region) or have a breakpoint distal of this and within YAC 810f11 (Huang et al. 1997; Wandstrat et al. 1998). Because the vast majority of the larger markers share this latter breakpoint, and given the high frequency of these, the findings suggest the possible presence of another duplicon or recombination-promoting sequence. Interestingly, the larger inv dup(15) are all de novo and exclusively maternal in origin.
Three phenotypes associated with the presence of a supernumerary marker chromosome derived from chromosome 15 are as follows: normal, the inv dup(15) syndrome, or PWS/AS. The inv dup(15) syndrome refers to a consistent phenotype of mental and developmental retardation, hypotonia, behavior disturbances, seizures, abnormal dermatoglyphics, and mild somatic abnormalities (Wisniewski et al. 1979; Leana-Cox et al. 1994). It is associated with the presence of the PWS/AS critical region as seen in the larger markers described above (Leana-Cox et al. 1994). The smaller markers (type 1 and type 2) are usually associated with a normal phenotype and may be familial, although some have been associated with a PWS or AS phenotype (Robinson et al. 1993). The latter are due to uniparental disomy of the normal chromosomes 15 (Robinson et al. 1993).
Although less common, direct duplications of proximal 15q11–15q13 have been observed (Pettigrew et al. 1987; Clayton-Smith et al. 1993a;Browne et al. 1997; Cook et al. 1997, 1998; Mohandas et al. 1999), some with the same breakpoints as PWS/AS deletions, whereas others have a more distal breakpoint similar to the large inv dup(15) (Repetto et al. 1998; Robinson et al. 1998). Patients with maternal duplications that include at least the PWS/AS critical region show developmental delay, or autism (Browne et al. 1997; Cook et al. 1997, 1998; Repetto et al. 1998), whereas a few individuals with paternal duplications have a nonspecific developmental delay (Mohandas et al. 1999). Finally, a few triplication cases have been reported (Schinzel et al. 1994; Cassidy et al. 1996; Long et al. 1998; Robinson et al. 1998), as well as paracentric inversions of 15q11–15q13 (Clayton-Smith et al. 1993b). It remains unclear whether these rearrangements are also mediated by homologous recombination of different copies of the END repeat duplicon or whether several different duplicons exist in 15q and are involved in the various rearrangements.
Other Examples of Genomic Disorders and Chromosomal Duplicons
Beside the examples discussed above, several other diseases are also caused by common DNA rearrangements. Proximal spinal muscular atrophy (SMA), a recessive-lethal condition affecting 1/10,000 births, is associated in ∼18% of cases with rearrangements in an inverted duplication of ∼500 kb in 5q13 (Melki et al. 1994; Lefebvre et al. 1995; Scharf et al. 1998). A second example is the recessive Hunter syndrome, due to mutations of the iduronate-2-sulfatase (IDS) gene in Xq28. Twenty percent of Hunter syndrome cases involve an inversion that disrupts the IDS gene, caused by homologous recombination between IDS and an IDS pseudogene 90 kb downstream (Bondeson et al. 1995). Although less well characterized, most cases of juvenile nephronophthisis (NPH) involve deletion of a 250-kb region on chromosome 2q13, flanked by repeat sequences (Konrad et al. 1996). Lastly, similar chromosome rearrangement events that are benign also occur in the genome. In studying a rare condition (Emery-Dreifuss muscular dystrophy, Xq28), Small et al (1997)identified a common 48-kb inversion that is present in the heterozygous state in 33% of females, arising from homologous recombination between flanking 11.3 kb duplicons of >99% sequence identity. As the human genome sequence is completed, the structure and role in chromosome rearrangements of these and other region-specific duplicons, including those currently unrecognized, will likely emerge.
Transchromosomal Duplicons
In addition to the chromosome-specific duplicons described above, a large number of genomic segments have been identified in the last few years that map to multiple non-homologous chromosomes (termed transchromosomal duplicons) (Borden et al. 1990; Wong et al. 1990;Buiting et al. 1992; Bernardi et al. 1993; Tomlinson et al. 1994;Eichler et al. 1996, 1997, 1999; Frippiat et al. 1997;Kehrer-Sawatzki et al. 1997; Regnier et al. 1997; Zimonjic et al. 1997;Eichler, 1998; Trask et al. 1998a,b; Brand-Arpon et al. 1999; Grewal et al. 1999; Horvath et al. 2000). Two regions of the human genome that seem unusually enriched for such transchromosomal duplication events are the pericentromeric (defined as the first Giemsa light or dark band flanking the primary constriction) and subtelomeric portions of chromosomes (Eichler 1998; Trask et al. 1998a). Detailed analyses of pericentromeric DNA in 10q11, 16p11, and 2p11 (Jackson et al. 1999a;Loftus et al. 1999; Horvath et al. 2000; J. Horvath, S. Schwartz, E.E. Eichler, unpubl.), for example, have identified zones of genomic duplicons located distal to α-satellite DNA. These regions are composed almost entirely of genic/pseudogenic segments that have been transposed from diverse regions of the hominoid genome. As one example, analysis of a 225-kb pericentromeric BAC sequence from chromosome 16p11 identified four different duplicons, including portions of the creatine-transporter, HERC2, variable κ, and variable heavy chain immunoglobulin genes (Loftus et al. 1999). All of these segments have been duplicated and transposed to 16p11 independently from their ancestral loci in Xq28, 15q13, 2p11, and 14q32, respectively. Comparative mapping and phylogenetic analysis of various pericentromeric segments reveals that they began to transpose within the hominoid genome relatively recently [estimates range from 3 to 20 million years ago (mya)]. Consequently, many of the paralogous segments share a high degree of sequence similarity (∼93%–98%). Furthermore, differences in map position and copy number have been observed among closely related primates (chimpanzee, human, and gorilla). Population studies of subtelomeric duplications have identified differences among different human ethnic groups (Trask et al. 1998a), suggesting that dynamic spread of these segments may be an ongoing process in the evolution of the human genome.
The molecular mechanism underlying the duplication and transposition of genomic segments to the pericentromeric or subtelomeric regions of chromosomes is not well understood. However, analysis of sequences located at the presumptive sites of insertion within the pericentromeric regions have uncovered several unusual classes of GC-rich repeats located in close (<1 kb) proximity to the boundaries of paralogy (Borden et al. 1990; Eichler et al., 1996, 1997; Eichler 1999). One class of these repeats (CAGGG; Fig. 4) localizes specifically to the pericentromeric regions of several different human autosomes, with an evolutionary origin predating the arrival of the duplicons themselves (Eichler et al. 1999). Moreover, the sequences bear some similarity both in sequence and organization to χ-recombination elements and immunoglobulin switch-recombination signal sequences, which are capable of mediating recombination events among non-allelic sequences. These repeats may play a role in pericentromeric duplications, by serving as a preferred transposition integration signal for the duplicons. It is interesting that similar GC-rich elements have also been mapped within the vicinity of recent subtelomeric duplications (Amann et al. 1996; Chute et al. 1997). In addition to these initial transposition seeding events within pericentromeric DNA, it is now clear that secondary events transfer larger segments of the duplicons among other pericentromeric regions (Fig. 4)—a process that has been termed pericentromeric swapping (Horvath et al. 2000; J. Horvath, S. Schwartz, and E.E. Eichler, unpubl.). The net effect of these duplication events is the generation of large (>100 kb) blocks of paralogy among multiple nonhomologous chromosomes, each of which is a mosaic of smaller duplicated segments (Fig. 4).
Schematic for duplicon formation and dispersal in chromosomes 15q13, 15q11, and 16p11.2. At top is shown BAC 778A2 (AC004583) spanning the 5′ half of the ancestral HERC2 gene, which has undergone intrachromosomal duplications to form multiple duplicons in 15q13 and 15q11 (see Fig. 3d). Miropeats comparison of repeat-masked sequence to a chromosome 16p11.2 BAC 17E1 (AC002041) illustrates a transchromosomal duplication and transposition of aHERC2-duplicon (Miropeats displays regions of similarity by joining lines between two sequences). Note the 27.1 kb ofHERC2-like sequence duplicated between these two segments (BESTFIT alignment indicates 94.7% genomic identity). The 17E1 BAC also identifies a second gene segment derived from an ancestral Xq28 locus (the creatine transporter gene, SLC6A8). These and several other locally duplicated segments (as denoted by arcs) form additional duplicons in 16p11. 17E1 also has 4 interspersed repeats including a large CAGGG-rich region (see text).
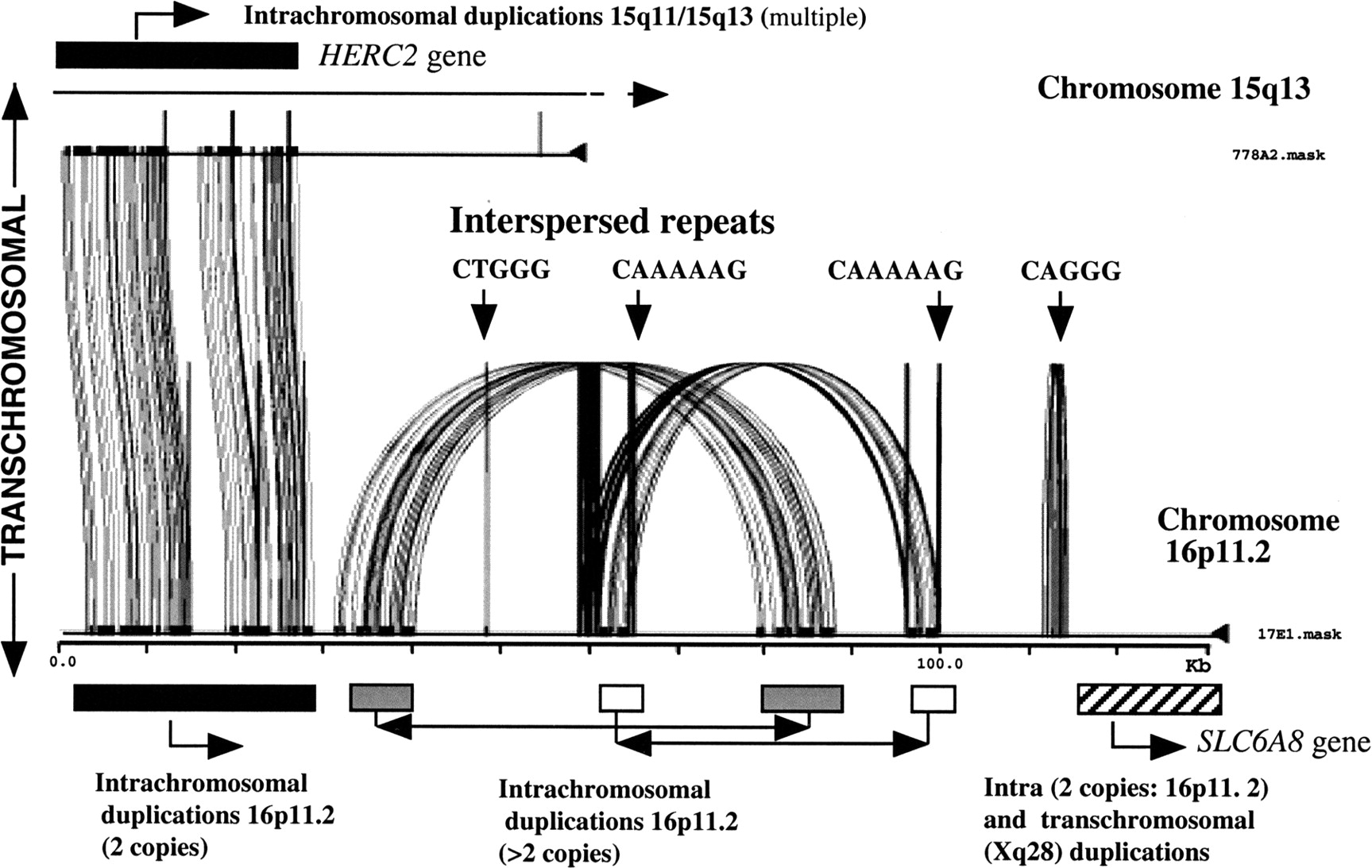
Is there a link between the dynamic duplicative nature of pericentromeric DNA and genomic duplicons mediating genomic disease? A direct connection is yet to be established, but there are some tantalizing associations. (1) CAGGG repeats have been mapped to several cytogenetic bands associated with recurrent chromosomal structural rearrangement and genomic disease (particularly within the proximal regions of 15q11 and 22q11) (Eichler et al. 1999). (2) There is a significant positional bias for duplicons and genomic diseases, as a group, to be near the pericentromeric region (e.g., 7q11.23, 15q11.2, 17p11.2, 17q11.2, and 22q11.2). Three of the most common rearrangements discussed above (SMS, PWS/AS, and VCFS), which involve complex duplicons, involve rearrangement between a duplicon situated near a proximal pericentromeric region and a duplicon located more distally. (3) Analyses of pericentromeric DNA on chromosome 10 (Jackson et al. 1999a,b), chromosome 22 (Dunham et al. 1999; Shaikh et al. 2000) and chromosome 16 (E.E. Eichler, unpubl.) suggest that intrachromosomal (chromosome-specific) duplicons become more prevalent as one approaches unique DNA. Furthermore, analysis of DNA from regions of pericentromeric DNA from 16p11.2 and 15q11 have identifiedHERC2 duplicons, derived from an ancestral locus in 15q13, as one component in a series of duplications that constitute 16p11 (Fig.4; Amos-Landgraf et al. 1999; Ji et al 1999, 2000; Loftus et al. 1999). These data suggest that the processes responsible for transchromosomal and intrachromosomal pericentromeric duplication and for disease-causing rearrangements may be molecularly interrelated. One scenario may be that pericentromeric DNA has created a favorable evolutionary milieu for the accumulation and transfer of duplicated segments. Recurrent chromosomal structural rearrangement associated with disease then arises due to the fact that both donor and acceptor recombining loci flank unique gene regions that are haplo-insufficient. Of course, not all duplicon-mediated rearrangements can be described as pericentromeric, nor is the process of recent genomic duplication restricted to pericentromeric and subtelomeric DNA (DeSilva et al. 1999; Loftus et al. 1999). The process of pericentromeric duplication, therefore, is likely one of several different mechanisms capable of creating paralogous sequences that can mediate genomic instability.
Conclusions and Perspectives
From studies discussed above and by others (Purandare and Patel 1997; Mazzarella and Schlessinger 1998; Lupski 1998), it is clear that the presence of duplicons predisposes to abnormal pairing and unequal crossing-over, leading to chromosome rearrangements in specific regions of the human genome. Combined, at least 0.7–1 per 1000 births will have a genomic disorder due to a de novo rearrangement between duplicons, attesting to the impact on medical care and economic burden. Deletions, duplications, inversions, translocations, and supernumerary marker chromosomes can all result from duplicon-mediated homologous recombination, suggesting this disease mechanism is a general phenomenon. The identification of novel junction fragments in most patients with CMT1A/HNPP, WS, SMS, duplication 17p11.2, and some 22q11.2 rearrangements, supports the idea that precise recombination events are associated with rearrangements involving even complex duplicons. It is likely that double-strand breaks might be the initiating event for these homologous recombination events (Lupski 1998), although it will be necessary to isolate the breakpoints from multiple patients in each class of disorders to more accurately model the mechanism of recombination.
Three general features of these duplicons can be proposed. (1) Large-size and high-sequence identity are important characteristics. The duplicons span at least thousands of base pairs to several hundred kilobases. The identity between the two recombining duplicons can be as high as 99.9% (int22h). Recombination hot spots are usually in regions of identical nucleotide sequence, of at least 200 to ∼450 bp in length, although this has not yet been examined in complex duplicon rearrangements. When genomic disorders are examined according to the distance between the flanking duplicons, it appears that the length of the duplicon increases as the distance between the duplicons increases (Lupski 1998). Large duplicons with high sequence identity may be necessary to initiate pairing and/or stabilize the recombination complex, for unequal crossing-over and recombination to occur. (2) Recombination promoting features may be present within duplicons. Most characterized duplicons contain one or more genes/pseudogenes. Two of the loci (VCX and HERC2) are expressed in germ cells, although it is unknown whether these or genes in other duplicons are transcribed in meiotic or post-meiotic germ cells, in which unequal crossover must occur. In yeast, double-strand breaks have been found primarily in promoter regions, suggesting a role for an open chromatin structure in homologous recombination (Osman and Subramani 1998). The presence of a sequence in SMS–REP that is homologous to one with autonomously replicating activity also suggests a role for open chromatin structure, and/or DNA replication. Sequences related to those that can promote recombination have been identified in recombination hot spots in CMT1A–REP and 22q11 rearrangements, and VNTRs are found within each copy of the S232 and LCR22 repeats, as well as within the END repeats (Ji et al. 2000). (3) In cases in which more than two duplicons exist in the same chromosome region, there are preferences as to which copies are involved in specific rearrangements. Further studies are needed to understand why some copies are chosen over others, but this may relate to sequence identity, orientation of the duplicons, presence of recombination promoting sequences, or chromatin structure.
Although much has been learned about the mechanisms for genomic disorders, many questions remain. (1) Some rearrangements occur almost exclusively in paternal meiosis (CMT1A duplication and F8 inversion), whereas others happen equally often in male and female meiosis, despite normal meiotic recombination being twice as frequent in females as in males. It has been suggested that there are male factors for, or female protection against, aberrant homologous recombination (Purandare and Patel 1997). One such factor may be the continued proliferation of spermatogonia after puberty, which may lead to a much greater likelihood of a structural rearrangement eventually occurring in sperm (Chandley 1991). (2) Most cases of genomic disorders are deletions, whereas duplications and inversions seem to be less common. This may arise from the mechanisms producing inter- or intra-chromosomal events between direct repeats (Fig. 1). A bias of ascertainment may also contribute, because deletions usually cause a more severe phenotype. (3) It is not known why most of these chromosome rearrangements involve duplications near pericentromeric regions (Eichler 1998), but as discussed above this may relate to the propensity for duplicons to cluster in these regions and for duplicons to generally disperse to adjacent chromosomal positions. Also not known is how duplicons several megabases apart search for and recombine with each other, nor what role higher order chromosome structures play in the process. (4) The origin and dispersal of various duplicons appears to be limited to primates. Although the CMT1A–REP seems to be a human- and chimpanzee-specific repeat (Kiyosawa and Chance 1996; Keller et al. 1999), preliminary studies on the END repeats suggests an origin of ∼15–20 mya (Christian et al. 1999; Ji et al. 1999), and the LCR22 duplicons arose at least 20–25 mya on the basis of their presence in Old World monkeys (Shaikh et al. 2000). Thus, the age of the duplicons appears to correlate with their complexity and the frequency of chromosome rearrangements. A comparative genomic study of the various duplicons first identified in the human genome will determine their evolutionary origin and provide insight into the mechanisms of formation and rearrangement of these duplicons.
Finally, it is possible that environmental factors may contribute to the chromosome rearrangement events that lead to genomic disorders. Preliminary studies suggest a link between paternal hydrocarbon exposure and PWS (Strakowski and Butler 1987; Cassidy et al. 1989;Åkefeldt et al. 1995), but other genomic disorders have not been examined. Several studies show an increased rate of mitotic homologous recombination between duplicated DNA segments. X-rays and other carcinogens were shown to significantly increase the reversion frequency of the pink-eyed unstable (pun ) mutation, a 70-kb internal tandem duplication of p (Schiestl et al. 1994, 1997). Reversion of mutations caused by tandem duplication have also been found in yeast, Chinese hamster and human cell lines, and were drastically increased by carcinogen treatments (Schiestl et al. 1988; Zhang and Jenssen 1992; Aubrecht et al., 1995). These events are all mediated by intrachromosomal recombination. It will be necessary and important to perform molecular and epidemiological studies to determine whether environmental factors (such as hydrocarbons) increase germ-line chromosome rearrangements that lead to the various genomic disorders in human. Given the remarkably high frequency of genomic disorders, as a group, if environmental risk factors were recognized it would allow proper measures to be taken to prevent preconceptional exposure, and thereby would significantly reduce the frequency as well as the economic and social costs of genomic disorders.
We thank James Amos-Landgraf for the original art of Figure 1. Work on duplicons in the author's laboratories is funded by a Neuromuscular Disease Research Program Grant from the Muscular Dystrophy Association (R.D.N.), by NIH grant GM58815–01 (E.E.E.), and by a Basil O'Connor Scholar award from the March of Dimes Birth Defects Foundation to E.E.E. (FY98–0712).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Notes
[1] Present address: Genentech, Inc., Department of Bioinformatics, South San Francisco, California 94080 USA.
[2] Corresponding author.
Notes
[3] E-MAIL [email protected]; FAX (216) 368-3432.
REFERENCES
- ↵A. ÅkefeldtM. AnvretU. GrandellR. NordlinderC. Gillberg(1995) Parental exposure to hydrocarbons in Prader-Willi syndrome. Dev. Med. Child. Neurol. 37:1101–1109.
- ↵J. AmannM. ValentineV.J. KiddJ.M. Lahti(1996) Localization of chi1-related helicase genes to human chromosome regions 12p11 and 12p13: Similarity between parts of these genes and conserved human telomeric-associated DNA. Genomics 32:260–265.
- ↵J.M. Amos-LandgrafY. JiW. GottliebT. DepinetA. WandstradtS.B. CassidyD.J. DriscollP.K. RoganS. SchwartzR.D. Nicholls(1999) Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am. J. Hum. Genet. 65:370–386.
- ↵J. AubrechtR. RugoR.H. Schiestl(1995) Carcinogens induce intrachromosomal recombination in human cells. Carcinogenesis 16:2841–2846.
- ↵A. Baldini(1999) Is the genetic basis of DiGeorge syndrome in HAND? Nat. Genet. 21:246–247.
- ↵A. BallabioB. BardoniS. GuioliE. BaslerG. Camerino(1990) Two families of low-copy-number repeats are interspersed on Xp22.3: Implications for the high frequency of deletions in this region. Genomics 8:263–270.
- ↵A. BaumerF. DutlyD. BalmerM. RiegelT. TukelM. Krajewska-WalaseckA.A. Schinzel(1998) High level of unequal meiotic crossovers at the origin of the 22q11.2 and 7q11.23 deletions. Hum. Mol. Genet. 7:887–894.
- ↵F. BernardiP. PatracchiniD. GammatiM. PinottiC. SchwienbacherG. BalleriniG. Marchetti(1993) In-frame deletion of von Willebrand factor A domains in a dominant type of von Willebrand disease. Hum. Mol. Genet. 2:545–548.
- ↵I.P. BlairJ. NashM.J. GordonG.A. Nicholson(1996) Prevalence and origin of de novo duplications in Charcot-Marie-Tooth disease type 1A: First report of a de novo duplication with a maternal origin. Am. J. Hum. Genet. 58:472–476.
- ↵E. BlennowK. Brondum-NielsenH. TeleniusN.P. CarterU. KristofferssonE. HolmbergC. GillbergM. Nordenskjold(1995) Fifty probands with extra structurally abnormal chromosomes characterized by fluorescence in situ hybridization. Am. J. Med. Genet. 55:85–94.
- ↵M.L. BondesonN. DahlH. MalmgrenW.J. KleijerT. TonnesenB.M. CarlbergU. Pettersson(1995) Inversion of the IDS gene resulting from recombination with IDS-related sequences is a common cause of the Hunter syndrome. Hum. Mol. Genet. 4:615–621.
- ↵P. BordenR. JaenichenH. Zachau(1990) Structural features of transposed human Vk genes and implications for the mechanism of their transpositions. Nucleic Acids Res. 18:2101–2107.
- ↵S. BortF. MartínezF. Palau(1997) Prevalence and parental origin of de novo 1.5-Mb duplication in Charcot-Marie-Tooth disease type 1A. Am. J. Hum. Genet. 60:230–233.
- ↵V. Brand-ArponS. RouquierH. MassaP.J. de JongC. FerrazP.A. IoannouJ.G. DemailleB.J. TraskD. Giorgi(1999) A genomic region encompassing a cluster of olfactory receptor genes and a myosin light chain kinase (MYLK) gene is duplicated on human chromosome regions 3q13-q21 and 3p13. Genomics 56:98–110.
- ↵C.E. BrowneN.R. DennisE. MaherF.L. LongJ.C. NicholsonJ. SillibourneJ.C.K. Barber(1997) Inherited interstitial duplications of proximal 15q: Genotype-phenotype correlations. Am. J. Hum. Genet. 61:1342–1352.
- ↵K. BuitingV. GregerB.H. BrownsteinR.M. MohrI. VoiculescuA. WinterpachtB. ZabelB. Horsthemke(1992) A putative gene family in 15q11–13 and 16p11.2: possible implications for Prader-Willi and Angelman syndromes. Proc. Natl. Acad. Sci. 89:5457–5461.
- ↵K. BuitingS. GrossY. JiG. SengerR.D. NichollsB. Horsthemke(1998) Expressed copies of the MN7 (D15F37) gene family map close to the common deletion breakpoints in the Prader-Willi/Angelman syndromes. Cytogenet. Cell Genet. 81:247–253.
- ↵K. BuitingC. KornerB. UlrichE. WahleB. Horsthemke(1999) The human gene for the poly(A)-specific ribonuclease (PARN) maps to 16p13 and has a truncated copy in the Prader-Willi/Angelman syndrome region on 15q11-q13. Cytogenet. Cell Genet. 87:125–131.
- ↵C. CarlsonH. SirotkinR. PanditaR. GoldbergJ. McKieR. WadeyS.R. PatanjaliS.M. WeissmanK. Anyane-YeboaD. Warburton(1997) Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. Am. J. Hum. Genet. 61:620–629.
- ↵R. CarrozzoE. RossiS.L. ChristianK. KittikamronC. LivieriA. CorriasL. PucciA. FoisP. SimiL. Bosio(1997) Inter- and intrachromosomal rearrangements are both involved in the origin of 15q11-q13 deletions in Prader-Willi syndrome. Am. J. Hum. Genet. 61:228–231.
- ↵S.B. CassidyA.J. GaineyM.G. Butler(1989) Occupational hydrocarbon exposure among fathers of Prader-Willi syndrome patients with and without deletion of 15q. Am. J. Hum. Genet. 44:806–810.
- ↵Cassidy, S.B., J. Conroy, L. Becker, and S. Schwartz. 1996. Paternal triplication of 15q11-q13 in a hypotonic, developmentally delayed child without Prader-Willi or Angelman syndrome..
- ↵P.F. ChanceK.H. Fischbeck(1994) Molecular genetics of Charcot-Marie-Tooth disease and related neuropathies. Hum. Mol. Genet. 3:1503–1507, . Am. J. Med. Genet. 62: 206–207..
- ↵P.F. ChanceM.K. AldersonK.A. LeppigM.W. LenschN. MatsunamiB. SmithP.D. SwansonS.J. OdelbergC.M. DistecheT.D. Bird(1993) DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 72:143–151.
- ↵A.C. Chandley(1991) On the parental origin of de novo mutation in man. J. Med. Genet. 28:217–223.
- ↵K.S. ChenP. ManianT. KoeuthL. PotockiQ. ZhaoA.C. ChinaultC.C. LeeJ.R. Lupski(1997) Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat. Genet. 17:154–163.
- ↵S.D. ChengN.B. SpinnerE.H. ZackaiJ.H. Knoll(1994) Cytogenetic and molecular characterization of inverted duplicated chromosomes 15 from 11 patients. Am. J. Hum. Genet. 55:753–759.
- ↵S.L. ChristianW.P. RobinsonB. HuangA. MutiranguraM.R. LineM. NakaoU. SurtiA. ChakravartiD.H. Ledbetter(1995) Molecular characterization of two proximal deletion breakpoint regions in both Prader-Willi and Angelman syndrome patients. Am. J. Hum. Genet. 57:40–48.
- ↵S.L. ChristianJ.A. FantesS.K. MewbornB. HuangD.H. Ledbetter(1999) Large genomic duplicons map to sites of instability in the Prader-Willi/Angelman syndrome chromosome region (15q11-q13). Hum. Mol. Genet. 8:1025–1037.
- ↵I. ChuteY. LeT. AshleyM.J. Dobson(1997) The telomere-associated DNA from human chromosome 20p contains a pseudotelomere structure and shares sequences with the subtelomeric regions of 4q and 18p. Genomics 46:51–60.
- ↵J. Clayton-SmithT. WebbX.J. ChengM.E. PembreyS. Malcolm(1993a) Duplication of chromosome 15 in the region 15q11–13 in a patient with developmental delay and ataxia with similarities to Angelman syndrome. J. Med. Genet. 30:529–531.
- ↵J. Clayton-SmithD.J. DriscollM.F. WatersT. WebbT. AndrewsS. MalcolmM.E. PembreyR.D. Nicholls(1993b) Difference in methylation patterns within the D15S9 region of chromosome 15q11-q13 in first cousins with Angelman syndrome and Prader-Willi syndrome. Am. J. Med. Genet. 47:683–686.
- ↵E.H. Cook Jr.V. LindgrenB.J. LeventhalR. CourchesneA. LincolnC. ShulmanC. LordE. Courchesne(1997) Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am. J. Hum. Genet. 60:928–934.
- ↵E.H. Cook Jr.R.Y. CourchesneN.J. CoxC. LordD. GonenS.J. GuterA. LincolnK. NixR. HaasB.L. LeventhalE. Courchesne(1998) Linkage-disequilibrium mapping of autistic disorder, with 15q11–13 markers. Am. J. Hum. Genet. 62:1077–1083.
- ↵A. de la ChapelleR. HervaM. KoivistoP. Aula(1981) A deletion in chromosome 22 can cause DiGeorge syndrome. Hum. Genet. 57:253–256.
- ↵U. DeSilvaH. MassaB.J. TraskE.D. Green(1999) Comparative mapping of the region of human chromosome 7 deleted in Williams syndrome. Genome Res. 9:428–436.
- ↵M.O. DorschnerV.P. SybertM. WeaverB.A. PletcherK. Stephens(2000) NF1 microdeletion breakpoints are clustered at flanking repetitive sequences. Hum. Mol. Genet. 9:35–46.
- ↵I. DunhamN. ShimizuB.A. RoeS. ChissoeA.R. HuntJ.E. CollinsR. BruskiewichD.M. BeareM. ClampL.J. Smink(1999) The DNA sequence of human chromosome 22. Nature 402:489–495.
- ↵F. DutlyA. Schinzel(1996) Unequal interchromosomal rearrangements may result in elastin gene deletions causing the Williams-Beuren syndrome. Hum. Mol. Genet. 5:1893–1898.
- ↵L. EdelmannR.K. PanditaB.E. Morrow(1999a) Low-copy repeats mediate the common 3-Mb deletion in patients with Velo-cardio-facial syndrome. Am. J. Hum. Genet. 64:1076–1086.
- ↵L. EdelmannR.K. PanditaE. SpiteriB. FunkeR. GoldbergN. PalanisamyR.S. ChagantiE. MagenisR.J. ShprintzenB.E. Morrow(1999b) A common molecular basis for rearrangement disorders on chromosome 22q11. Hum. Mol. Genet. 8:1157–1167.
- ↵L. EdelmannE. SpiteriN. McCainR. GoldbergR.K. PanditaS. DuongJ. FoxD. BlumenthalS.R. LalaniL.G. ShafferB.E. Morrow(1999c) A common breakpoint on 11q23 in carriers of the constitutional t(11;22) translocation. Am. J. Hum. Genet. 65:1608–1616.
- ↵E.E. Eichler(1998) Masquerading repeats: Paralogous pitfalls of the human genome. Genome Res. 8:758–762.
- ↵(1999) Repetitive conundrums of centromere structure and function. Hum. Mol. Genet. 8:151–155, ibid.
- ↵E.E. EichlerF. LuY. ShenR. AntonacciV. JurecicN.A. DoggettR.K. MoyzisA. BaldiniR.A. GibbsD.L. Nelson(1996) Duplication of a gene-rich cluster between 16p11.1 and Xq28: A novel pericentromeric-directed mechanism for paralogous genome evolution. Hum. Mol. Genet. 5:899–912.
- ↵E.E. EichlerM.L. BudarfM. RocchiL.L. DeavenN.A. DoggettA. BaldiniD.L. NelsonH.W. Mohrenweiser(1997) Interchromosomal duplications of the adrenoleukodystrophy locus: A phenomenon of pericentromeric plasticity. Hum. Mol. Genet. 6:991–1002.
- ↵E.E. EichlerN. ArchidiaconoM. Rocchi(1999) CAGGG repeats and the pericentromeric duplication of the hominoid genome. Genome Res. 9:1048–1058.
- ↵A.K. EwartC.A. MorrisD. AtkinsonW. JinK. SternesP. SpalloneA.D. StockM. LeppertM.T. Keating(1993) Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat. Genet. 5:11–16.
- ↵U. Francke(1999) Williams-Beuren syndrome: genes and mechanisms. Hum. Mol. Genet. 8:1947–1954.
- ↵J.P. FrippiatP. DardS. MarshG. WinterM.P. Lefranc(1997) Immunoglobulin lamda light chain orphons on human chromosome 8q11.2. Eur. J. Immunol. 27:1260–1265.
- ↵B. FunkeL. EdelmannN. McCainR.K. PanditaJ. FerreiraS. MerscherM. ZohouriL. CannizzaroA. ShanskeB.E. Morrow(1999) Der(22) syndrome and velo-cardio-facial syndrome/DiGeorge syndrome share a 1.5-Mb region of overlap on chromosome 22q11. Am. J. Hum. Genet. 64:747–758.
- ↵F. GreenbergV. GuzzettaR. Montes de Oca-LunaR.E. MagenisA.C. SmithS.F. RichterI. KondoW.B. DobynsP.I. PatelJ.R. Lupski(1991) Molecular analysis of the Smith-Magenis syndrome: a possible contiguous-gene syndrome associated with del(17)(p11.2). Am. J. Hum. Genet. 49:1207–1218.
- ↵P.K. GrewalM. van GeelR.R. FrantsP. de JongJ.E. Hewitt(1999) Recent amplification of the human FRG1 gene during primate evolution. Gene 227:79–88.
- ↵E.L. HockenhullM.J. CaretteK. MetcalfeD. DonnaiA.P. ReadM. Tassabehji(1999) A complete physical contig and partial transcript map of the Williams syndrome critical region. Genomics 58:138–145.
- ↵J.E. HoogendijkG.W. HenselsA.A. Gabreels-FestenF.J. GabreelsE.A. JanssenP. de JongheJ.J. MartinC. van BroeckhovenL.J. ValentijnF. Baas(1992) De novo mutation in hereditary motor and sensory neuropathy type I. Lancet 339:1081–1082.
- ↵J.E. HorvathL. ViggianoB.J. LoftusM.D. AdamsN. ArchidiaconoM. RocchiE.E. Eichler(2000) Molecular structure and evolution of an alpha satellite/non-alpha satellite junction at 16p11. Hum. Mol. Genet. 9:113–123.
- ↵B. HuangJ.A. CrollaS.L. ChristianM.E. Wolf-LedbetterM.E. MachaP.N. PapenhausenD.H. Ledbetter(1997) Refined molecular characterization of the breakpoints in small inv dup(15) chromosomes. Hum. Genet. 99:11–17.
- ↵M.S. JacksonM. RocchiG. ThompsonT. HearnM. CrosierJ. GuyD. KirkL. MulliganA. RiccoS. Piccininni(1999a) Sequences flanking the centromere of human chromosome 10 are a complex patchwork of arm-specific sequences, stable duplications, and unstable sequences with homologies to telomeric and other centromeric locations. Hum. Mol. Genet. 8:205–215.
- ↵M. JacksonM. RocchiT. HearnM. CrosierJ. GuyL. ViggianoS. PiccininniA. RiccoR. MarzellaN. Archidiacono(1999b) Characterisation of the heterochromatin/euchromatin boundary at 10q11 and identification of novel transcripts by repeat induced instability. Am. J. Hum. Genet. 65:A56.
- ↵Y. JiM.J. WalkowiczK. BuitingD.K. JohnsonR.E. TarvinE.M. RinchikB. HorsthemkeL. StubbsR.D. Nicholls(1999) The ancestral gene for transcribed, low-copy repeats in the Prader-Willi/Angelman region encodes a large protein implicated in protein trafficking, which is deficient in mice with neuromuscular and spermiogenic abnormalities. Hum. Mol. Genet. 8:533–542.
- ↵Y. JiN.A. RebertJ.M. JoslinM.J. HigginsR.A. SchultzR.D. Nicholls(2000) Structure of the highly conserved HERC2 gene and of multiple partially duplicated paralogs in human. Genome Res. 10:319–329.
- ↵Y.H. JiangE. Lev-LehmanJ. BresslerT.F. TsaiA.L. Beaudet(1999) Genetics of Angelman syndrome. Am. J. Hum. Genet. 65:1–6.
- ↵R.C. JuyalL.E. FigueraX. HaugeS.H. ElseaJ.R. LupskiF. GreenbergA. BaldiniP.I. Patel(1996) Molecular analyses of 17p11.2 deletions in 62 Smith-Magenis syndrome patients. Am. J. Hum. Genet. 58:998–1007.
- ↵H. Kehrer-SawatzkiT. SchwickardtG. AssumG. RocchiW. Krone(1997) A third neurofibromatosis type 1 (NF1) pseudogene at chromosome 15q11.2. Hum. Genet. 100:595–600.
- ↵M.P. KellerB.A. SeifriedP.F. Chance(1999) Molecular evolution of the CMT1A-REP region: A human- and chimpanzee-specific repeat. Mol. Biol. Evol. 16:1019–1026.
- ↵H. KiyosawaP.F. Chance(1996) Primate origin of the CMT1A-REP repeat and analysis of a putative transposon-associated recombinational hot spot. Hum. Mol. Genet. 5:745–753.
- ↵H. KiyosawaM.W. LenschP.F. Chance(1995) Analysis of the CMT1A-REP repeat: mapping crossover breakpoints in CMT1A and HNPP. Hum. Mol. Genet. 4:2327–2334.
- ↵M. KonradS. SaunierL. HeidetF. SilbermannF. BenessyJ. CaladoD. Le PaslierM. BroyerM.C. GublerC. Antignac(1996) Large homozygous deletions of the 2q13 region are a major cause of juvenile nephronophthisis. Hum. Mol. Genet. 5:367–371.
- ↵B.T. LahnD.C. Page(2000) A human sex-chromosomal gene family expressed in male germ cells and encoding variably charged proteins. Hum. Mol. Genet. 9:311–319.
- ↵D. LakichH.H.Jr. KazazianS.E. AntonarakisJ. Gitschier(1993) Inversions disrupting the factor VIII gene as a common cause of severe haemophilia A. Nat. Genet. 5:236–241.
- ↵J. Leana-CoxL. JenkinsC.G. PalmerR. PlattnerL. SheppardW.L. FlejterJ. ZackowskiF. TsienS. Schwartz(1994) Molecular cytogenetic analysis of inv dup(15) chromosomes, using probes specific for the Prader-Willi/Angelman syndrome region: Clinical implications. Am. J. Hum. Genet. 54:748–756.
- ↵D.H. LedbetterV.M. RiccardiS.D. AirhartR.J. StrobelB.S. KeenanJ.D. Crawford(1981) Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. New. Engl. J. Med. 304:325–329.
- ↵S. LefebvreL. BurglenS. ReboulletO. ClermontP. BurletL. ViolletB. BenichouC. CruaudP. MillasseauM. Zeviani(1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80:155–165.
- ↵B. LevinsonS. KenwrickD. LakichG. Hammonds Jr.J. Gitschier(1990) A transcribed gene in an intron of the human factor VIII gene. Genomics 7:1–11.
- ↵X.-M. LiP. YenL. Shapiro(1992) Characterization of a low copy repetitive element S232 involved in the generation of frequent deletions of the distal short arm of the human X chromosome. Nucleic Acids Res. 20:1117–1122.
- ↵B.J. LoftusU.-J. KimV.P. SneddonF. KalushR. BrandonJ. FuhrmannT. MasonM. L. CrosbyM. BarnsteadL. Cronin(1999) Genome duplications and other features in 12 Mbp of DNA sequence from human chromosome 16p and 16q. Genomics 60:295–308.
- ↵F.L. LongD.P. DuckettL.J. BillamD.K. WilliamsJ.A. Crolla(1998) Triplication of 15q11-q13 with inv dup(15) in a female with developmental delay. J. Med. Genet. 5:425–428.
- ↵J. LopesE. LeGuernR. GouiderS. TardieuN. AbbasN. BiroukM. GugenheimP. BoucheY. AgidA. Brice(1996) Recombination hot spot in a 3.2-kb region of the Charcot-Marie-Tooth type 1A repeat sequences: New tools for molecular diagnosis of hereditary neuropathy with liability to pressure palsies and of Charcot-Marie-Tooth Type 1A. Am. J. Hum. Genet. 58:1223–1230.
- ↵J. LopesA. VandenbergheS. TardieuV. IonasescuN. LevyN. WoodN. TachiP. BoucheP. LatourA. BriceE. LeGuern(1997) Sex-dependent rearrangements resulting in CMT1A and HNPP. Nat. Genet. 17:136–137.
- ↵J. LopesN. RaviseA. VandenbergheF. PalauV. IonasescuM. MayerN. LevyN. WoodN. TachiP. Bouche(1998) Fine mapping of de novo CMT1A and HNPP rearrangements within CMT1A-REPs evidences two distinct sex-dependent mechanisms and candidate sequences involved in recombination. Hum. Mol. Genet. 7:141–148.
- ↵J.R. Lupski(1998) Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 14:417–422.
- ↵R. MazzarellaD. Schlessinger(1998) Pathological consequences of sequence duplications in the human genome. Genome Res. 8:1007–1021.
- ↵K.E. McTaggartM.L. BudarfD.A. DriscollB.S. EmanuelP. FerreiraH.E. McDermid(1998) Cat eye syndrome chromosome breakpoint clustering: Identification of two intervals also associated with 22q11 deletion syndrome breakpoints. Cytogenet. Cell Genet. 81:222–228.
- ↵J. MelkiS. LefebvreL. BurglenP. BurletO. ClermontP. MillasseauS. ReboulletB. BenichouM. ZevianiD. Le Paslier(1994) De novo and inherited deletions of the 5q13 region in spinal muscular atrophies. Science 264:1474–1477.
- ↵C. MignonP. MalzacA. MonclaD. DepetrisN. RoeckelM.F. CroquetteM.G. Mattei(1996) Clinical heterogeneity in 16 patients with inv dup 15 chromosomes: Cytogenetic and molecular studies, search for an imprinting effect. Eur. J. Hum. Genet. 4:88–100.
- ↵T.K. MohandasJ.P. ParkR.A. SpellmanJ.J. FilianoA.C. MamourianA.B. HawkD.R. BelloniW.W. NollJ.B. Moeschler(1999) Paternally derived de novo interstitial duplication of proximal 15q in a patient with developmental delay. Am. J. Med. Genet. 82:294–300.
- ↵C.A. MorrisI.T. ThomasF. Greenberg(1993) Williams syndrome: Autosomal dominant inheritance. Am. J. Med. Genet. 47:478–481.
- ↵J. NaylorA. BrinkeS. HassockP.M. GreenF. Giannelli(1993) Charateristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum. Mol. Genet. 2:1773–1778.
- ↵J.A. NaylorD. BuckP.M. GreenH. WilliamsonD. BentleyF. Giannelli(1995) Investigation of the factor VIII intron 22 repeated region (int22h) and the associated inversion junctions. Hum. Mol. Genet. 4:1217–1224.
- ↵J.A. NaylorP. NicholsonA. GoodeveS. HassockI. PeakeF. Giannelli(1996) A novel DNA inversion causing severe hemophilia A. Blood 87:3255–3261.
- ↵E. NelisC. Van BroeckhovenP. De JongheA. LofgrenA. VandenbergheP. LatourE. Le GuernA. BriceM.L. MostacciuoloF. Schiavon(1996) Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: A European collaborative study. Eur. J. Hum. Genet. 4:25–33.
- ↵R.D. NichollsS. SaitohB. Horsthemke(1998) Imprinting in Prader-Willi and Angelman syndromes. Trends Genet. 14:194–200.
- ↵E. NickersonF. GreenbergM.T. KeatingC. McCaskillL.G. Shaffer(1995) Deletions of the elastin gene at 7q11.23 occur in approximately 90% of patients with Williams syndrome. Am. J. Hum. Genet. 56:1156–1161.
- ↵L.R. OsborneJ. HerbrickT. GreavetteH.H.Q. HengL. TsuiS.W. Scherer(1997) PMS2-related genes flank the rearrangement breakpoints associated with Williams syndrome and other diseases on human chromosome 7. Genomics 45:402–406.
- ↵F. OsmanS. Subramani(1998) Double-strand break-induced recombination in eukaryotes. Prog. Nucleic Acid Res. Mol. Biol. 58:263–299.
- ↵F. PalauA. LofgrenP. De JongheS. BortE. NelisT. SevillaJ.J. MartinJ. VilchezF. PrietoC. Van Broeckhoven(1993) Origin of de novo duplication in Charcot-Marie-Tooth disease type 1A: Unequal nonsister chromatid exchange during spermatogenesis. Hum. Mol. Genet. 2:2031–2035.
- ↵P.I. PatelJ.R. Lupski(1994) Charcot-Marie-Tooth disease: A new paradigm for the mechanism of inherited disease. Trends Genet. 10:128–133.
- ↵L. PentaoC.A. WiseA.C. ChinaultP.I. PatelJ.R. Lupski(1992) Charcot-Marie-Tooth type 1A tandem duplication appears to arise from recombination at repeat sequences flanking the 1.5 Mb monomer unit. Nat. Genet. 2:292–300.
- ↵L.A. Perez JuradoR. PeoplesP. KaplanB.C. HamelU. Francke(1996) Molecular definition of the chromosome 7 deletion in Williams syndrome and parent-of-origin effects on growth. Am. J. Hum. Genet. 59:781–792.
- ↵L.A. Perez JuradoY. WangR. PeoplesA. ColomaJ. CrucesU. Francke(1998) A duplicated gene in the breakpoint regions of the 7q11.23 Williams-Beuren syndrome deletion encodes the initiator binding protein TFII-I and BAP-135, a phosphorylation target of BTK. Hum. Mol. Genet. 7:325–334.
- ↵A.L. PettigrewS.M. GollinF. GreenbergV.M. RiccardiD.H. Ledbetter(1987) Duplication of proximal 15q as a cause of Prader-Willi syndrome. Am. J. Med. Genet. 28:791–802.
- ↵L. PotockiK-S. ChenS-S. ParkD.E. OsterholmM.A. WithersV. KimonisA.M. SummersW.S. MeschinoK. Anyane-YeboaC.D. Kashork(2000) Molecular mechanism for duplication 17p11.2 – the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat. Genet. 24:84–87.
- ↵S.M. PurandareP.I. Patel(1997) Recombination hot spots and human disease. Genome Res. 7:773–786.
- ↵V. RegnierM. MeddebG. LecointreF. RichardA. DuvergerV.C. NguyenB. DutrillauxA. BernheimG. Danglot(1997) Emergence and scattering of multiple neurofibromatosis (NF1)-related sequences during hominoid evolution suggest a process of pericentromeric interchromosomal transposition. Hum. Mol. Genet. 6:9–16.
- ↵L.T. ReiterT. MurakamiT. KoeuthL. PentaoD.M. MuznyR.A. GibbsJ.R. Lupski(1996) A recombination hot spot responsible for two inherited peripheral neuropathies is located near a mariner transposon-like element. Nat. Genet. 12:288–297.
- ↵L.T. ReiterT. MurakamiT. KoeuthR.A. GibbsJ.R. Lupski(1997) The human COX10 gene is disrupted during homologous recombination between the 24 kb proximal and distal CMT1A-REPs. Hum. Mol. Genet. 6:1595–1603.
- ↵L.T. ReiterP.J. HastingsE. NelisP. De JongheC. Van BroeckhovenJ.R. Lupski(1998) Human meiotic recombination products revealed by sequencing a hot spot for homologous strand exchange in multiple HNPP deletion patients. Am. J. Hum. Genet. 62:1023–1033.
- ↵G.M. RepettoL.M. WhiteP.J. BaderD. JohnsonJ.H.M. Knoll(1998) Interstitial duplications of chromosome region 15q11q13: Clinical and molecular characterization. Am. J. Med. Genet. 9:82–89.
- ↵W.P. RobinsonJ. WagstaffF. BernasconiC. BaccichettiL. ArtifoniE. FranzoniL. SuslakL.Y. ShihH. AvivA.A. Schinzel(1993) Uniparental disomy explains the occurrence of the Angelman or Prader-Willi syndrome in patients with an additional small inv dup(15) chromosome. J. Med. Genet. 30:756–760.
- ↵W.P. RobinsonJ. WaslynkaF. BernasconiM. WangS. ClarkD. KotzotA. Schinzel(1996) Delineation of 7q11.2 deletions associated with Williams-Beuren syndrome and mapping of a repetitive sequence to within and to either side of the common deletion. Genomics 34:17–23.
- ↵W.P. RobinsonF. DutlyR.D. NichollsF. BernasconiM. PenaherreraR.C. MichaelisD. AbeliovichA.A. Schinzel(1998) The mechanisms involved in formation of deletions and duplications of 15q11-q13. J. Med. Genet. 35:130–136.
- ↵J.P. RossiterM. YoungM.L. KimberlandP. HutterR.P. KetterlingJ. GitschierJ. HorstM.A. MorrisD. SchaidP. de Moerloose(1994) Factor VIII gene inversions causing severe hemophilia A originate almost exclusively in male cells. Hum. Mol. Genet. 3:1035–1039.
- ↵L.S. SadlerL.K. RobinsonK.R. VerdaasdonkR. Gingell(1993) The Williams syndrome: Evidence for possible autosomal dominant inheritance. Am. J. Med. Genet. 47:468–470.
- ↵J.M. ScharfM.G. EndrizziA. WetterS. HuangT.G. ThompsonK. ZerresW.F. DietrichB. WirthL.M. Kunkel(1998) Identification of a candidate modifying gene for spinal muscular atrophy by comparative genomics. Nat. Genet. 20:83–86.
- ↵R.H. SchiestlS. IgarashiP.J. Hastings(1988) Analysis of the mechanism for reversion of a disrupted gene. Genetics 119:237–247.
- ↵R.H. SchiestlF. KhogaliN. Carls(1994) Reversion of the mouse pink-eyed unstable mutation induced by low doses of X-rays. Science 266:1573–1576.
- ↵R.H. SchiestlJ. AubrechtF. KhogaliN. Carls(1997) Carcinogens induce reversion of the mouse pink-eyed unstable mutation. Proc. Natl. Acad. Sci. 94:4576–4581.
- ↵A. SchinzelW. SchmidM. FraccaroL. TiepoloO. ZuffardiJ.M. OpitzJ. LindstenP. ZetterqvistH. EnellC. Baccichetti(1981) The ‘cat eye syndrome‘: Dicentric small marker chromosome probably derived from a no.22 (tetrasomy 22pter to q11) associated with a characteristic phenotype. Report of 11 patients and delineation of the clinical picture. Hum. Genet. 57:148–158.
- ↵A.A. SchinzelL. BrecevicF. BernasconiF. BinkertF. BerthetA. WuilloudW.P. Robinson(1994) Intrachromosomal triplication of 15q11-q13. J. Med. Genet. 31:798–803.
- ↵S. SchwartzT.W. Depinet(1996) Studies of ‘acentric’ and ‘dicentric’ marker chromosomes: Implications for the definition of the functional centromere. Am. J. Hum. Genet. (Suppl.) 59:A14.
- ↵T.H. ShaikhH. KurahashiS.C. SaittaA. Mizrahy O'HareP. HuA.A. RoeD.A. DriscollD.M. McDonald-McGinnE.H. ZackaiM.L. BudarfB.S. Emanuel(2000) Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: Genomic organization and deletion endpoint analysis. Hum. Mol. Genet. 9:489–501.
- ↵K. SmallJ. IberS.T. Warren(1997) Emerin deletion reveals a common X-chromosome inversion mediated by inverted repeats. Nat. Genet. 16:96–99.
- ↵J.M. SoucieB. EvattD. Jackson(1998) Occurrence of hemophilia in the United States. Am. J. Hematol. 59:288–294, Hemophilia Surveillance System Project Investigators.
- ↵S.M. StrakowskiM.G. Butler(1987) Paternal hydrocarbon exposure in Prader-Willi syndrome. Lancet 8573:1458.
- ↵I.M. TomlinsonG.P. CookN.P. CarterR. ElaswarapuS. SmithG. WalterL. BuluwelaT.H. RabbittsG. Winter(1994) Human immunoglobulin VH and D segments on chromosomes 15q11.2 and 16p11.2. Hum. Mol. Genet. 3:853–860.
- ↵B. TraskC. FriedmanA. Martin-GallardoL. RowenC. AkinbamiJ. BlankenshipC. CollinsD. GiorgiS. IadonatoF. Johnson(1998a) Members of the olfactory receptor gene family are contained in large blocks of DNA duplicated polymorphically near the ends of human chromosomes. Hum. Mol. Genet. 7:13–26.
- ↵B.J. TraskH. MassaV. Brand-ArponK. ChanC. FriedmanO.T. NguyenE.E. EichlerG. van den EnghS. RouquierH. ShizuyaD. Giorgi(1998b) Large multi-chromosomal duplications encompass many members of the olfactory receptor gene family in the human genome. Hum. Mol. Genet. 7:2007–2020.
- ↵A.S. WaldmanR.M. Liskay(1988) Dependence of intrachromosomal recombination in mammalian cells on uninterrupted homology. Mol. Cell. Biol. 8:5350–5357.
- ↵A.E. WandstratJ. Leana-CoxL. JenkinsS. Schwartz(1998) Molecular cytogenetic evidence for a common breakpoint in the largest inverted duplications of chromosome 15. Am. J. Hum. Genet. 62:925–936.
- ↵M.S. WangA. SchinzelD. KotzotD. BalmerR. CaseyB.N. ChodirkerJ. GyftodimouM.B. PetersenE. Lopez-RangelW.P. Robinson(1999) Molecular and clinical correlation study of Williams-Beuren syndrome: No evidence of molecular factors in the deletion region or imprinting affecting clinical outcome. Am. J. Med. Genet. 86:34–43.
- ↵D.I. WilsonJ. BurnP. ScamblerJ. Goodship(1993) DiGeorge syndrome: Part of CATCH 22. J. Med. Genet. 30:852–856.
- ↵C.A. WiseC.A. GarciaS.N. DavisZ. HejuL. PentaoP.I. PatelJ.R. Lupski(1993) Molecular analyses of unrelated Charcot-Marie-Tooth (CMT) disease patients suggest a high frequency of the CMTIA duplication. Am. J. Hum. Genet. 53:853–863.
- ↵L. WisniewskiT. HassoldJ. HeffelfingerJ.V. Higgins(1979) Cytogenetic and clinical studies in five cases of inv dup(15). Hum. Genet. 50:259–270.
- ↵Z. WongN. RoyleA. Jeffreys(1990) A novel human DNA polymorphism resulting from transfer of DNA from chromosome 6 to chromosome 16. Genomics 7:222–234.
- ↵C. WuP. FriedlanderC. LamoureuxM. Zannis-HadjopoulosG.B. Price(1993) cDNA clones contain autonomous replication activity. Biochim. Biophys. Acta 1174:241–257.
- ↵H. YamagishiV. GargR. MatsuokaT. ThomasD. Srivastava(1999) A molecular pathway revealing a genetic basis for human cardiac and craniofacial defects. Science 283:1158–1161.
- ↵P.H. YenX.M. LiS.P. TsaiC. JohnsonT. MohandasL.J. Shapiro(1990) Frequent deletions of the human X chromosome distal short arm result from recombination between low copy repetitive elements. Cell 61:603–610.
- ↵L.H. ZhangD. Jenssen(1992) Reversion of the hprt mutant clone SP5 by intrachromosomal recombination. Carcinogenesis 13:609–615.
- ↵D. ZimonjicM. KelleyJ. RubinS. AaronsonN. Popescu(1997) Fluorescence in situ hybridization analysis of keratinocyte growth factor gene amplification and dispersion in evolution of great apes and humans. Proc. Natl. Acad. Sci. 94:11461–11465.