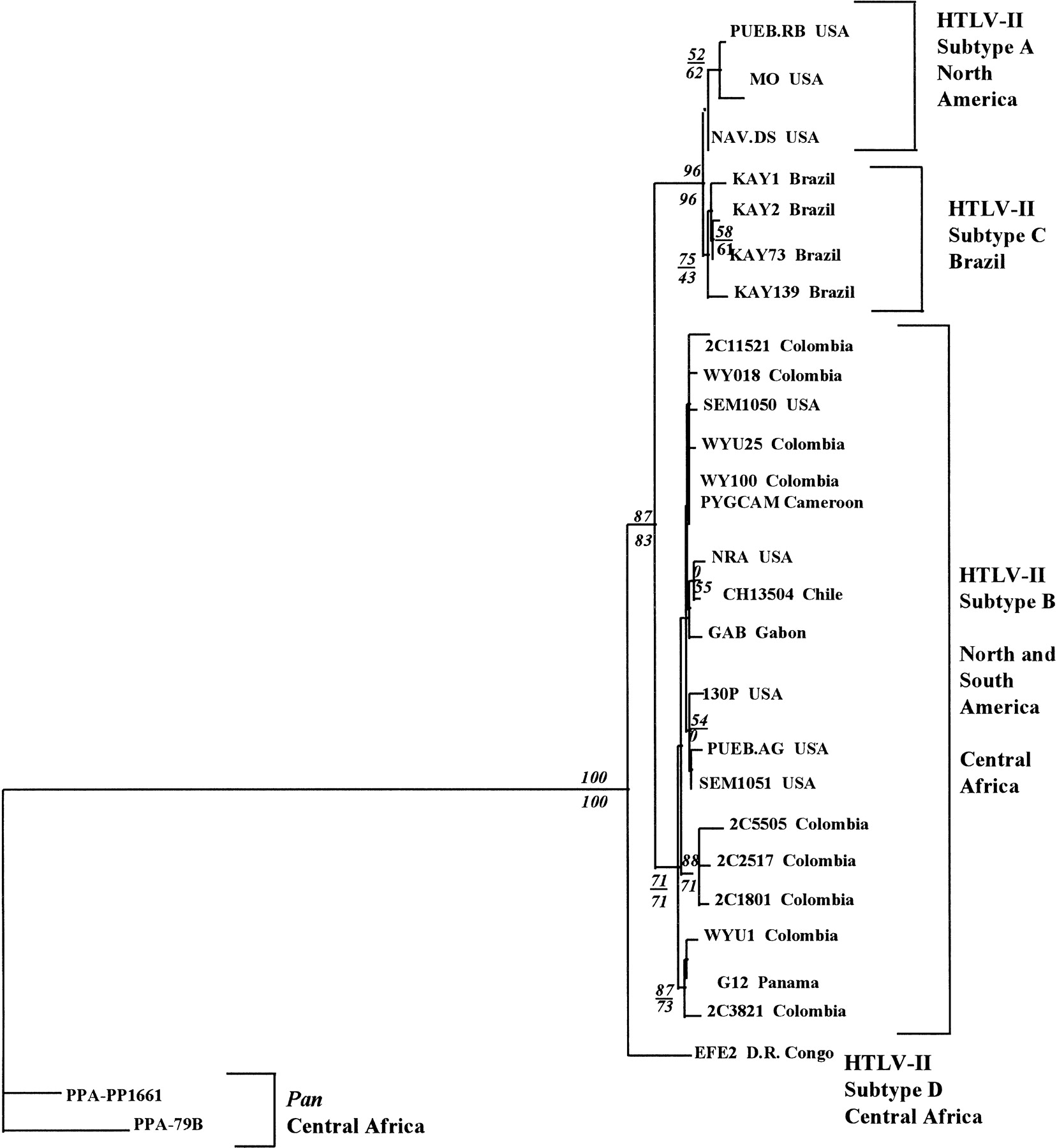
Figure 5.
Phylogenetic tree based on 417 bp alignment of LTR region sequenced from 28 viral strains of HTLV-II/STLV-II excluding IVDU. Shown is one of three equivalent trees obtained by ME estimated by NJ. Corroborating trees were obtained by MP that yielded a 50% majority rule of 15 trees of equivalent length = 209; C.I. = 0.856 and ML (ln likelihood = −1595.1; 2776 trees examined). Numbers in italics denote bootstrap proportions in support of adjacent node (NJ/MP). Identical conditions presented in phylogenetic analyses from Fig. 4were used.