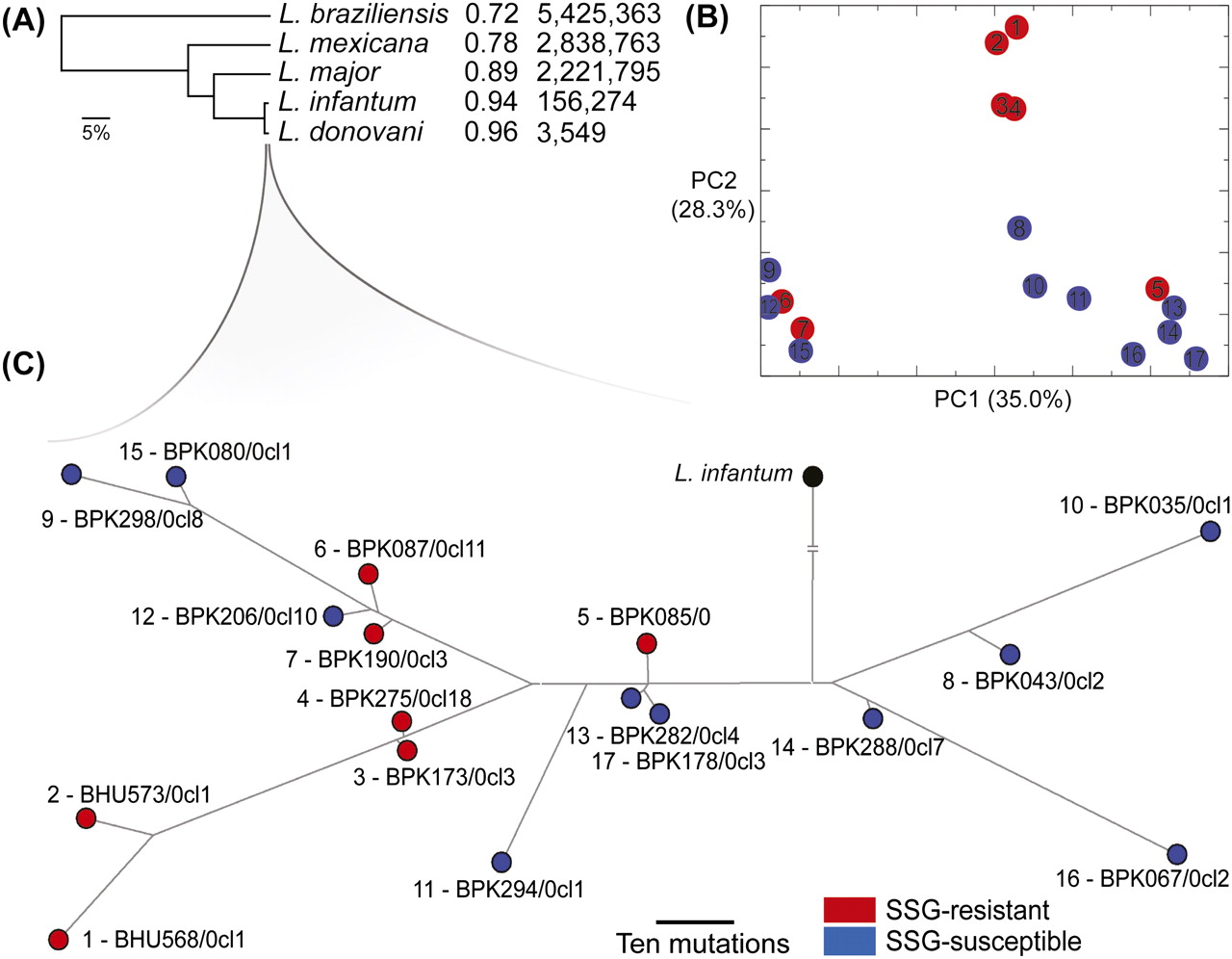
The phylogenomic context of functional variation in clinical L. donovani lines. (A) A neighbor-joining phylogenomic tree of Leishmania species. The relative similarity is shown (1% was equivalent to 108,000 mutations). The first column indicates the fraction of the known genomes orthologously aligned to L. donovani and the second is the number of SNPs identified (Table 2). (B) Principle component analysis (PCA) of genomic SNP variation in the 17 L. donovani lines resistant (red) and susceptible (blue) to SGG. The two most significant PCs distinguished three main groups, accounting for 63.3% of the total variation. The PCA L. donovani line numbers correspond to those shown in C, a median-joining phylogenetic network of genome-wide nonsynonymous sites variation (396 SNPs) for samples resistant (red) and susceptible (blue) to SSG from India (beginning with BHU) and Nepal (BPK). The branch lengths displayed are proportional to the number of differences between lines. The position of the ancestral node for L. infantum (black) was shortened: For comparison, the L. donovani:L. infantum branch length was 138 times that between the most divergent L. donovani (1, BHU568/0cl1; and 10, BPK035/0cl1).