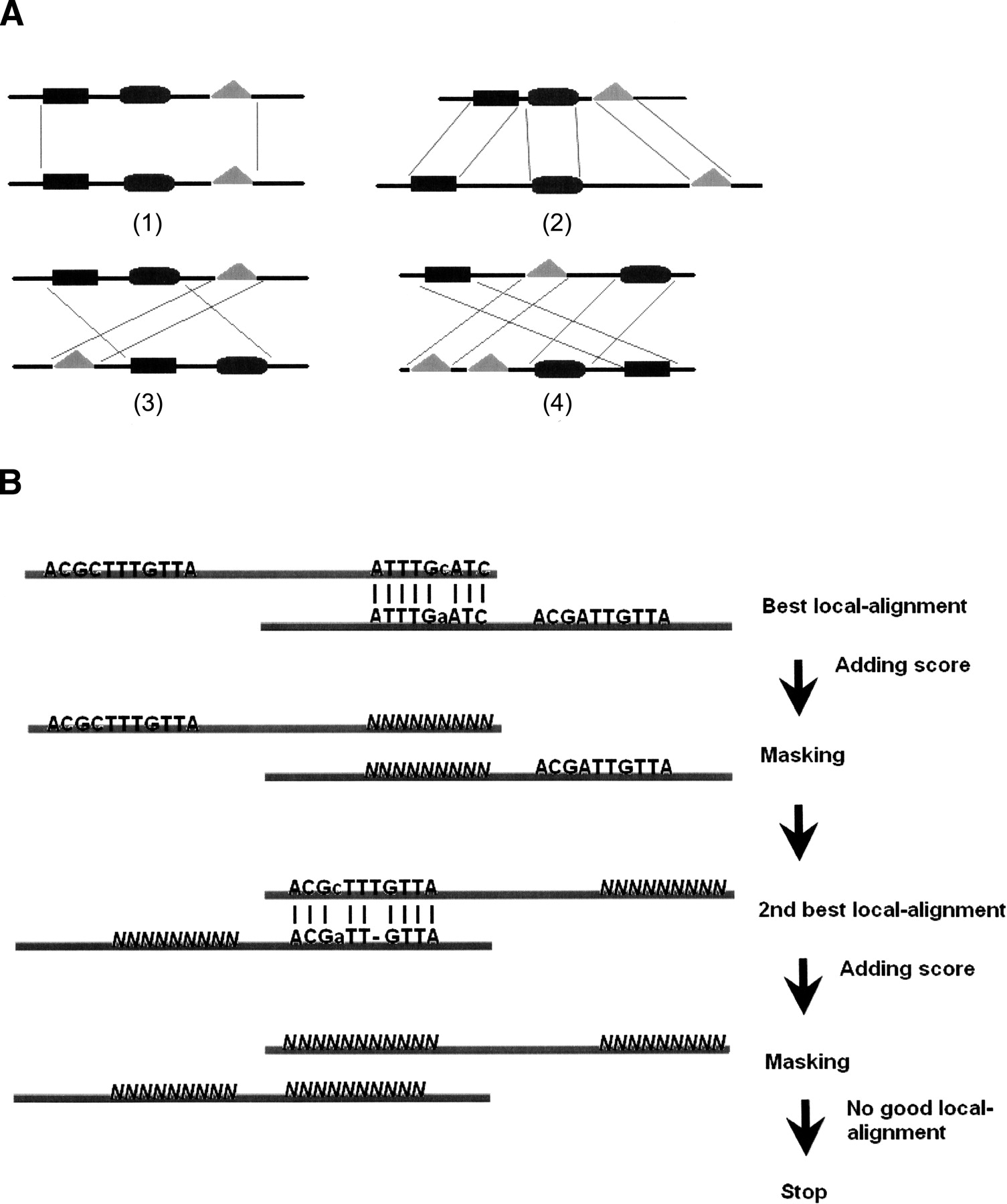
Module-Alignment. (A) An illustration of three pairs of orthologous CRMs: All three CRM pairs consist of TFBSs generated from the same motifs (squares, ellipses, and triangles). (1) Orthologous CRMs with conserved number, order, and distances. (2–4) Orthologous CRMs with different distances, order, and number of TFBSs. (B) Workflow of Module-Alignment: Module-Alignment iteratively performs local alignment and masks out conserved regions. The mutations and gaps between the alignable segments on the upper and lower sequences incur severe penalty so that Smith-Waterman can detect only the best local alignment in the first row. The conservation score from local alignment is the score of the best local alignment. However, the conservation score from Module-Alignment is the sum of the two local alignments scores from the two alignable sequence segments. Module-alignment is not designed to align any two sequences with any arbitrary lengths. Its input sequences should be potentially orthologous CRMs with lengths of several dozen to several hundred base pairs.