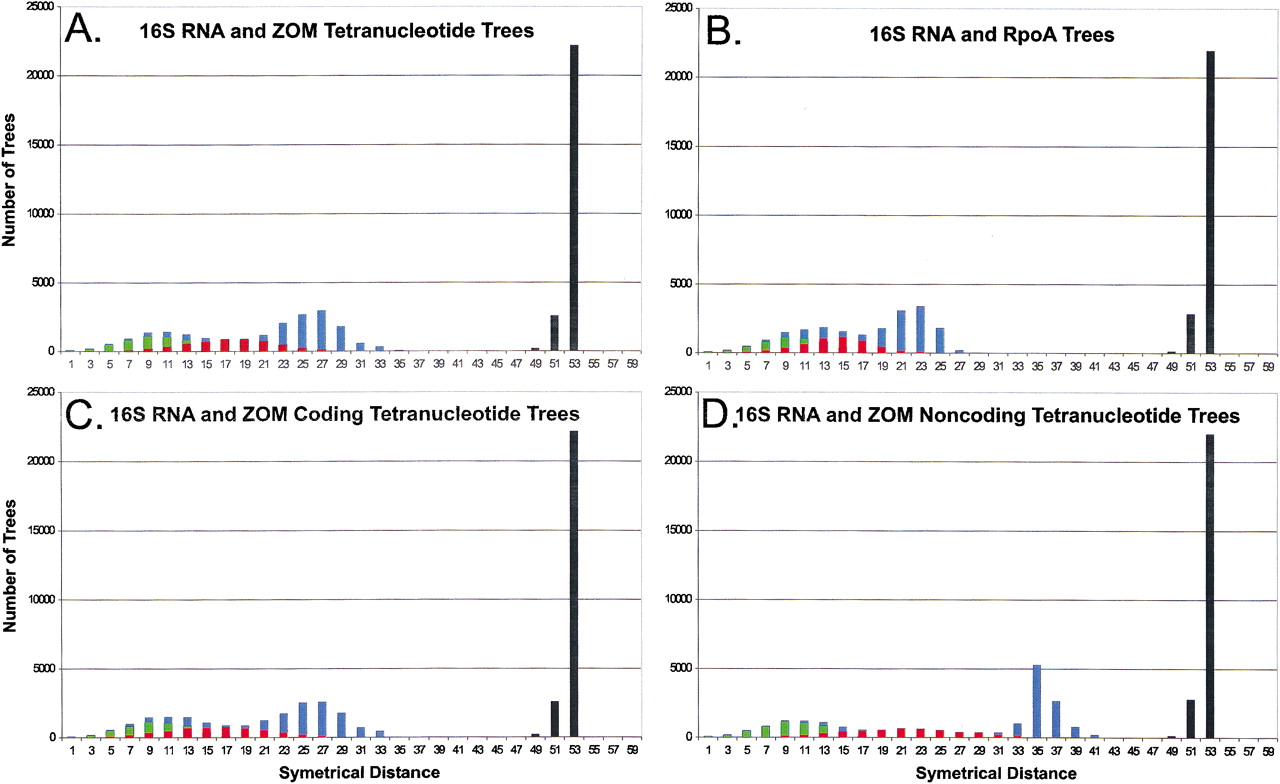
Tree distance analysis of phylogenies of 27 prokaryotes. One hundred phylogenies were created using bootstrapping techniques for these organisms based on 16S rRNA or RpoA sequences, or tetranucleotide usage deviation (TUD). Tree distances were determined using symmetrical parameters (Penny and Hendy 1985) using Paup 4.0b8 (Swofford 1998). (A–D) The distances between each set of phylogenetic trees; black columns represent all comparisons with random trees. Tree comparisons represented are: (A) 16S rRNA and tetranucleotide trees based on zero-order Markov criteria (green, 16S rRNA; red, tetranucleotide; blue, 16S vs. tetranucleotide); (B) 16S rRNA and RpoA trees (green, 16S rRNA; red, RpoA; blue, 16S vs. RpoA); (C) 16S rRNA and coding DNA tetranucleotide trees based on zero-order Markov criteria (green, 16S rRNA; red, coding DNA tetranucleotide; blue, 16S vs. coding DNA tetranucleotide); (D) 16S rRNA and noncoding DNA tetranucleotide trees based on zero-order Markov criteria (green, 16S rRNA; red, noncoding DNA tetranucleotide; blue, 16S vs. noncoding DNA tetranucleotide).