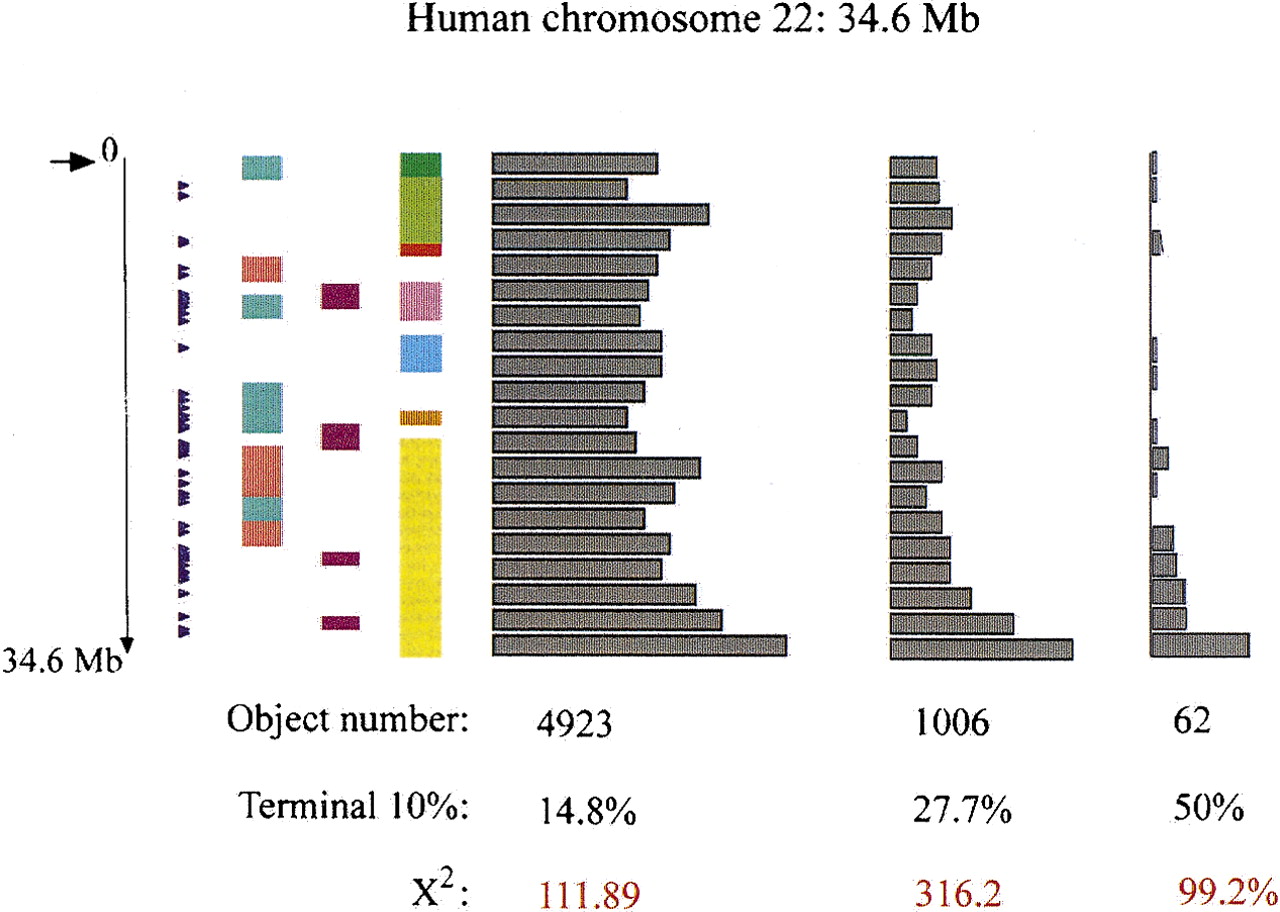
Distribution of tandem repeats corresponding to different queries along human chromosome 22. Tandem repeats have been identified within the human chromosome 22 sequence using the Tandem Repeats Finder (TRF) software with the following options: alignment parameters = (2,3,5), minimum alignment score to report repeat = 50, maximum period size = 500. Redundancy was then eliminated, and Alu and satellite sequences (152 were identified) were filtered. The arrow (top left) shows the centromere position. The position of the 51 chr22 Genethon microsatellites present in the database is shown with arrow heads. GC-rich (pink) or -poor (green) areas, regions of increased recombination, and known mouse synteny correspondence are as indicated in Dunham et al. (1999) Distributions obtained with different queries:
(A, Left) U > = 6, N > = 3
(B, Middle) U > 16, N > = 3, L > 100
(C, Right) U > 16, N > = 3, %GC > = 65%, BGC > = 0.3, %M > = 85% (U = unit length, N = copy number, L = total length, %GC = GC percent, BGC = G/C bias = ..%G-%C../(%G + %C), %M (percent matches) is the average similarity of each motif with the consensus motif). Percentages reported correspond to the proportion of objects in the last 10% of total length. χ2 values were calculated by comparing the last 10% of the chromosome with the mean number of objects along the whole chromosome. χ2 threshold of significance (homogeneity hypothesis is rejected if χ2 is greater than threshold) is 3.841 with P = 5%, and 10.827 with P = 0.1% (1 degree of freedom).