
A DNA Polymorphism Discovery Resource for Research on Human Genetic Variation
Identifying the genes conferring susceptibility or resistance to common human diseases should become increasingly feasible with improved methods for finding DNA sequence variants on a genome-wide scale (Collins et al. 1997; Landegren et al. 1998; Wang et al. 1998). To facilitate the discovery of DNA sequence variants, the National Human Genome Research Institute (NHGRI) of NIH, working with the Centers for Disease Control and Prevention, the National Institute of Environmental Health Sciences, and several individual investigators, has assembled a DNA Polymorphism Discovery Resource of samples from 450 U.S. residents with ancestry from all the major regions of the world. This DNA Polymorphism Discovery Resource will be immensely valuable for the discovery of human genetic variation, which other follow-up studies can relate to health and disease.
Most successes so far in finding genes that contribute to disease risk have been for highly penetrant diseases caused by single genes, such as cystic fibrosis (Kerem et al. 1989; Rommens et al. 1989). To locate genes affecting these rare disorders, researchers perform linkage analysis on families, which requires 300–500 highly informative genetic markers spanning the entire human genome. However, it has been considerably harder to locate the genes contributing to the risk of common diseases such as diabetes, heart disease, cancers, and psychiatric disorders, because these phenotypes are affected by multiple genes, each with small effect; environmental contributions are also important. Instead of linkage analysis on families it may be much more efficient to perform association analysis on many affected and unaffected individuals, which would require hundreds of thousands of variants spread over the entire genome (Risch and Merikangas 1996). Such a large number of variants is currently not available. The DNA Polymorphism Discovery Resource is designed to promote their discovery.
About 90% of sequence variants in humans are differences in single bases of DNA, called single nucleotide polymorphisms (SNPs). SNPs in the coding regions of genes (cSNPs) or in regulatory regions are more likely to cause functional differences than SNPs elsewhere. Although most SNPs do not affect gene function, a large number of mapped SNPs will be valuable as markers throughout the genome for finding SNPs that do affect gene function, as linkage disequilibrium over tens to hundreds of kilobases is expected to be found in many regions of the human genome. Both SNPs and cSNPs can be identified by using the DNA Polymorphism Discovery Resource.
When two random chromosomes are compared, they differ at ∼
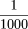 nucleotides (Kwok et al. 1996). When all chromosomes from 40 individuals are screened, about 17 million SNPs are expected to be found, out of the 3 billion
bases in human DNA. Only a small proportion of these SNPs are expected to be in coding regions, as coding regions are ∼5%
of the genome and are less likely to have SNPs (Nickerson et al. 1998). Thus the number of cSNPs is estimated to be ∼500,000, an average of about 6 per gene.
nucleotides (Kwok et al. 1996). When all chromosomes from 40 individuals are screened, about 17 million SNPs are expected to be found, out of the 3 billion
bases in human DNA. Only a small proportion of these SNPs are expected to be in coding regions, as coding regions are ∼5%
of the genome and are less likely to have SNPs (Nickerson et al. 1998). Thus the number of cSNPs is estimated to be ∼500,000, an average of about 6 per gene.
There is thus widespread interest in finding SNPs. They are also more numerous, more stable, and potentially easier to score than the microsatellite repeat variants currently used for mapping genes. Given the large potential role of SNPs in human disease it is important to have these data in the public domain (Heller and Eisenberg 1998). Consequently NIH recently began an initiative for large-scale discovery of SNPs and for improving the technology to detect them. For this initiative, as well as others that are just starting, a common, geographically diverse DNA resource for SNP discovery is crucial, so that researchers can benefit from the cumulative power gained by sharing a common resource, analogous to the demonstrated benefits of the common CEPH panel for genetic mapping and the RH panels for physical mapping. Accumulating information on the same samples will allow validation, quality control, and comparisons of different methods of detecting SNPs.
The DNA Polymorphism Discovery Resource has been assembled to serve this function. It includes cell lines and DNA from 450 anonymous, unrelated individuals, with equal numbers of females and males. No medical, phenotypic, or ethnicity information is included. There are predefined subsets with 8, 24, 44, and 90 samples as well as the complete set, with each subset containing the smaller subsets. The subsets have the same range of diversity as the complete set. This span of sample sizes will allow researchers to use common sets of samples that are of the appropriate size to address various questions. Fewer samples will be needed when researchers are developing new technologies or looking for common variants; however, a more extensive set of samples will be needed when researchers are scaling up technologies or looking for rarer variants.
The sampling strategy for the DNA Polymorphism Discovery Resource facilitates finding genetic variants in the entire human population (Table 1). Any population contains ∼85% of the worldwide genetic variation, but none contains all of it (Barbujani et al. 1997). Because a random sample of U.S. residents would include genomes of mostly European origin, the DNA Polymorphism Discovery Resource includes individuals with non-European ancestry at more than their frequency in the U.S. population, although no attempt was made to be exhaustive or precisely balanced. The individuals sampled are U.S. residents who have ancestors from the major geographic regions of the world—Europe, Africa, the Americas, and Asia (Weiss 1998). Many U.S. residents have ancestors from more than one region, and such individuals are included in the DNA Polymorphism Discovery Resource. The European–American group includes non-Hispanic whites; the African–American group includes non-Hispanic blacks; the Americas group includes Mexican–Americans and Native Americans; and the Asian–American group includes individuals whose ancestors came from several countries in East and South Asia.
Composition of the DNA Polymorphism Discovery Resource
Table 1 shows the number of individuals sampled from each population group and the expected number of genomes corresponding to the proportion of their ancestry from each geographic region. The second column shows current estimates of the average amounts of admixture for the groups sampled: African admixture for the European–Americans, Native American and African admixture for the Mexican–Americans, and European admixture for the other groups (Hanis et al. 1991; Parra et al. 1998).
All samples came from individuals who gave informed consent explicitly to be part of this DNA Polymorphism Discovery Resource. The consent forms explain that the purpose of the DNA Polymorphism Discovery Resource is to discover genetic variation. All collection of consents and samples received Institutional Review Board (IRB) approval. For the Native American samples, consent was obtained first from the tribe and then from individuals; in addition, community issues were discussed with other Native Americans and representatives of the Indian Health Service.
To ensure a diverse collection, information on geographic origin and gender was collected for each individual sampled, but once the DNA Polymorphism Discovery Resource was assembled, all identifying and demographic information was removed from the individual samples. Because 52% more individuals gave consent than were included in the DNA Polymorphism Discovery Resource, no one, not even those sampled, knows which individuals are actually included. A summary of the number of individuals sampled from each group is available for the complete collection and the predefined subsets, but no identifiers are associated with individual samples.
The DNA Polymorphism Discovery Resource was designed to be used to discover variants in human DNA, not to assess the frequency of variants in particular groups. Thus, the DNA Polymorphism Discovery Resource is not useful for population-specific medical or anthropological studies, such as the Human Genome Diversity Project (Committee on Human Genome Diversity 1997). The lack of information on geographic origin and phenotype may be seen by some as a lost opportunity for research. However, many ethical issues are raised by including such information, and addressing these issues will take time. The Ethical, Legal, and Social Implications research program of NHGRI has made these issues a high priority for consideration; they include avoidance of stigmatization, population definition, community consultation, and education about the meaning of genetic variation. These issues must be addressed so that any potential harmful effects of research on human genetic variation are minimized while the benefits are made widely available. Delay in the development of the DNA Polymorphism Discovery Resource while these issues were being addressed would have resulted in lost research opportunities, so the admittedly limited approach described here was adopted as an initial step in this area.
A concern has been raised that researchers might try to identify the ethnicity of the samples (Foster and Freeman 1998). This probably cannot be done precisely, as most variants occur in most populations, and some individuals have ancestry from multiple regions. Nonetheless, the Native Americans agreed to participate only on the condition that such identification would not be attempted. Consequently, attempting to define the population affiliation of the samples in the DNA Polymorphism Discovery Resource is a violation of the consent form and thus a violation of regulation 45 CFR 46 for the protection of human subjects. Users will be required to agree not to attempt to identify ethnicity when they obtain samples and when they access the data derived from the DNA Polymorphism Discovery Resource in the NIH SNP database (see below).
The samples are available from the Coriell Institute for Medical Research as part of the National Institute of General Medical Sciences Human Genetic Mutant Cell Repository (http://umdnj.edu/locus/nigms/). The NHGRI, in consultation with the Office for Protection from Research Risks, considers the material in the DNA Polymorphism Discovery Resource to be in the category of human subjects, for which exemption 4, use of anonymous samples, should apply. All use of material in the DNA Polymorphism Discovery Resource must be reviewed by an IRB and approved or designated as exempt. The Coriell Institute requires that researchers submit their proposed research plans so it can check that the plans are in accordance with the consent forms signed by the individuals who agreed to participate in the DNA Polymorphism Discovery Resource.
The NIH National Center for Biotechnology Information has created a database to collect information on all SNPs found for each sample in the DNA Polymorphism Discovery Resource, as part of their SNP database, dbSNP (http://www.ncbi.nlm.nih.gov/SNP/). SNP discoverers are encouraged to submit their data to dbSNP because a central database will give researchers ready access to data on the variants found using the DNA Polymorphism Discovery Resource as well as the ability to verify the SNP data and to find associations among SNPs.
Finding genetic variants on a large scale is a natural extension of the Human Genome Project and has become one of its new goals (Collins et al. 1998). At least 100,000 SNPs are expected to be discovered in the next 3 years and placed in the public domain. These variants will allow researchers to identify the genetic basis for common diseases and to improve the efficacy of drugs and other therapies.
Acknowledgments
We particularly thank the individuals who agreed to participate in the DNA Polymorphism Discovery Resource and the research groups who provided the samples. We thank the participants in the DNA Polymorphism Discovery Resource planning meeting, held at the NIH on December 8–9, 1997, which laid out the blueprint for the plan described here: Kenneth Buetow, Linda Burhansstipanov, Aravinda Chakravarti, Georgia Dunston, Jonathan Friedlaender, Bronya Keats, Charles Langley, Andrew Merriwether, John Moore, Robert Nussbaum, Madison Powers, Nancy Press, Edward Sondik, Karen Steinberg, Diane Wagener, LeRoy Walters, Bruce Weir, and Kenneth Weiss. We also thank Jeanne Beck, Christine Beiswanger, Jean Findlay, William Freeman, Bettie Graham, Judith Greenberg, Mark Guyer, Clifford Johnson, Robert Johnson, Elke Jordan, Cay Loria, Jennifer Madans, Geraldine McQuillan, Glenn Pinder, Edward Sondik, Elizabeth Thomson, and Lorraine Toji for assistance with the project. We thank the National Institute of General Medical Sciences for supporting this project.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL brooks{at}exchange.nih.gov; FAX (301) 480-2770.
- Cold Spring Harbor Laboratory Press








